
卵巢癌中线粒体转录终止因子1的表达及其意义
2018-04-09 03:37自加吉孙美涛王唯斯陈莹戴莉萍余敏熊伟
生物技术通讯 2018年2期
自加吉,孙美涛,王唯斯,陈莹,戴莉萍,余敏,熊伟
1.大理大学 基础医学院,云南 大理 671000;2.大理州第一人民医院 病理科,云南 大理671000;3.云南大学 生命科学学院,云南 昆明 650091
卵巢癌是女性生殖系统三大恶性肿瘤之一,病死率居妇科恶性肿瘤的首位,且发病率逐年增加[1]。虽然近年发现了很多新的治疗方式,但其预后仍未得到明显改善[2]。目前,诊断卵巢癌发生发展的分子标记物仍然十分有限。人线粒体转录终止因子1(mitochondrial transcription termi⁃nationfactor1,MTERF1)基因定位于染色体7q21.2,包含4个外显子,其编码的蛋白由399个氨基酸残基构成,具有3个亮氨酸拉链结构和2个独立的DNA结合区[3]。MTERF1蛋白与线粒体DNA(mitochondrialDNA,mtDNA)上编 码 16S rRNA基因与tRNALeu(UUR)基因分界处的一段28 bp的序列特异结合,从而终止mtDNA重链的转录[4]。对人宫颈癌HeLa细胞株的体外研究发现,MTERF1蛋白正调控肿瘤细胞的线粒体基因表达和氧化磷酸化水平,进而促进肿瘤细胞的增殖,提示MTERF1在肿瘤细胞的发生发展中发挥作用[5]。然而,MTERF1基因在人卵巢癌发生发展中的作用机制仍不清楚。以基因芯片或转录组分析为代表的高通量测序目前已成为癌症研究的强大工具,但由于样本数量、测序平台及分析方法的不同,常导致实验结果有偏差,分析结果比较片面。如何有效利用上述资源,是当前科研人员面临的重要问题。数据整合分析可联合多种数据消除以上因素的影响,从整体角度来理解这些数据,因而生物数据整合分析逐渐成为大数据挖掘的重要方向,目前已经广泛应用于多种类型的癌症分析中[6]。Oncomine数据库是大型肿瘤基因芯片整合数据库,最新数据涵盖已知所有的癌症类型,包含715个数据集,共计86 733个癌组织及正常组织数据。利用Oncomine数据库可以进行肿瘤及其相应的正常组织中的表达差异分析,寻找潜在感兴趣的基因,其整合的大量数据保证分析结果具有极大的参考价值,也可以发现新的肿瘤标记物和基因治疗靶点。
1 材料与方法
1.1 从Oncomine数据库中挖掘数据
本实验中,我们在Oncomine数据库设定的筛选条件为:①Gene:MTERF1;②Analysis type:can⁃cervs.normalanalysis;③Cancertype:ovarian cancer;④Data type:mRNA;⑤Sample type:clini⁃cal specimen;⑥临界值设定条件:P<0.0001,fold change>2.0,gene rank=top 10%。
1.2 Real time RT-PCR检测MTERF1 mRNA在卵巢癌和正常卵巢上皮组织中的表达
选取2016年大理州第一人民医院病理科收集的10例卵巢癌组织及其对应的正常卵巢上皮组织,提取各组织的总RNA,逆转录合成cDNA后,调整各组间cDNA浓度一致,用CFX96荧光定量PCR扩增仪分别扩增目的基因和内参基因,目的基因和内参基因引物序列见表1。反应条件:95℃预变性30 s;95℃变性10 s,58℃退火30 s,72℃延伸1 min,35个循环;最后72延伸10 min。每个样品设置3个复孔,所有实验重复3次。根据内参的标准化计算目的基因的相对表达量,即


表1 目的基因和内参基因的引物序列
计算卵巢癌组织中MTERF1mRNA水平与其对应正常卵巢上皮组织的mRNA水平的比值。
1.3 Western印迹检测MTERF1蛋白在卵巢癌和正常卵巢上皮组织中的表达
提取10例卵巢癌及其配对正常卵巢上皮组织总蛋白,采用BCA法测定各组蛋白质的浓度。取40 μg蛋白质上样,SDS-PAGE分离总蛋白,将蛋白质转印至PVDF膜上,用5%的脱脂牛奶封闭2 h,加入1∶1000稀释的兔抗人MTERF1单克隆抗体,4℃冰箱内孵育过夜,内参为GAPDH;TBST液洗涤3次,每次5 min,加入1∶2000稀释的羊抗兔二抗,4℃孵育2 h;TBST洗涤3次,每次5 min。将超敏型ECL显影液滴于PVDF膜条上,用ImageQuant LAS500超灵敏化学发光成像系统显色曝光,扫描成图像。采用Image J 1.46软件对蛋白质条带进行灰度值分析,计算MTERF1/GAP⁃DH灰度值比值。
1.4 Kaplan-Meier plotter分析患者生存周期
Kaplan-Meier plotter数据库含有从基因表达汇编(Gene Expression Omnibus,GEO)数据库中下载的包括mRNA表达和临床资料的1657例卵巢癌病例信息。利用在线数据库(http://kmplot. com/analysis/)的卵巢癌数据集对MTERF1进行分析,得到相应的Kaplan-Meier生存曲线、风险比(hazard ratio,HR)及Log rankP(Pvalue)值。
1.4.1MTERF1表达水平与卵巢癌患者预后的关系 探针选择为JetSet best probe set,所需有效Affymetrix ID为204871_at(MTERF1)。首先根据MTERF1mRNA水平分为低表达组和高表达组,截尾数据为癌症无进展生存期(progression-freesurvival,PFS),符合条件的总病例数为1436例,分析MTERF1表达水平与卵巢癌预后的关系;然后截尾数据为总生存期(overall survival,OS),符合条件总病例数为1657例,分析MTERF1表达水平与卵巢癌预后的关系。筛选条件为:①Cancer:Ovarian cancer;②Gene:MTERF1;③Survival:PFS or OS。
1.4.2MTERF1表达对不同分期卵巢癌患者预后的影响 探针选择为JetSet best probe set,然后分别设截尾数据为OS和PFS,所需有效Affyme⁃trix ID为204871_at(MTERF1),在Stage选项下分别选择1期、1+2期、2期、2+3期、2+3+4期、3期、3+4期、4期,在线分析MTERF1表达对不同分期卵巢癌患者预后的影响。
1.5 统计学分析
正常卵巢组织和卵巢癌病例组之间MTERF1表达的差异采用t检验。所有统计学分析采用SPSS 22.0软件进行,以P<0.05为差异有统计学意义。
2 结果
2.1 Oncomine数据库分析MTERF1在卵巢癌中的表达
自2001年始,Oncomine数据库中共有8个研究涉及MTERF1在卵巢癌组织和正常卵巢组织中的表达,包括2211个样本[7-12]。荟萃8个研究结果发现,与对照组相比,人MTERF1在卵巢癌中高表达,其中位数为3867.0,其高表达具有显著性差异(P=0.042)。Oncomine数据库中有4个基因芯片数据集研究的统计分析结果,显示卵巢癌中MTERF1mRNA高表达(图1)。
2.2 卵巢癌组织与正常卵巢上皮组织中MTERF1 mRNA的表达
Real time RT-PCR检测10例卵巢癌及其配对正常卵巢上皮组织中MTERF1mRNA表达水平,结果显示6例卵巢癌组织的MTERF1mRNA水平较正常卵巢上皮组织显著增高(P<0.05),4例卵巢癌组织中增高不显著(P>0.05)(图2)。
2.3 卵巢癌组织与正常卵巢上皮组织MTERF1蛋白的表达
Western印迹检测10例配对卵巢癌与正常卵巢上皮组织中MTERF1蛋白的表达水平,结果同样显示6例卵巢癌组织中MTERF1蛋白水平较正常卵巢上皮组织显著增高(P<0.05),4例卵巢癌组织中增高不显著(P>0.05),与mRNA表达水平的检测结果一致(图3)。
2.4 卵巢癌患者的预后与MTERF1 mRNA表达量的关系

图1 Oncomine数据库部分所选队列中MTERF1基因在卵巢癌中的表达研究结果A:Yoshihara等(P<0.001);B:TCGA(P=0.004);C:Adib等(P=0.256);D:Lu等(P=0.026)
采用Kaplan-Meier plotter数据库进行生存周期分析、风险比和Log-Rank检验,进一步明确MTERF1基因表达量与卵巢癌病人预后的关系。当截尾数据设置为卵巢癌患者PFS,数据库中符合条件的病例总数为1436例,且涵盖各个分期的卵巢癌病例。经Kaplan-Meier plotter数据库在线分析,HR=1.22,log-rankP值为0.002。当截尾数据设置为卵巢癌患者OS,数据库中符合条件的病例总数为1657例,Kaplan-Meier Plotter数据库在线分析,HR=1.12,Log rankP值为0.091(图4)。
2.5 各分期卵巢癌患者预后与MTERF1 mRNA水平的关系
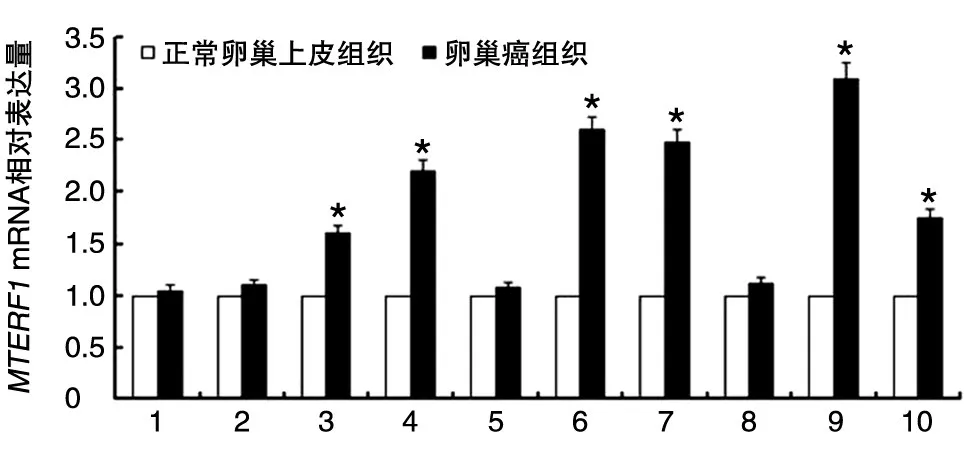
图2 10例卵巢癌与其相应的正常卵巢上皮组织中MTERF1 mRNA表达水平的比值(*P<0.05)
截尾数据为PFS时,早期卵巢癌(1、1+2或2期)中MTERF1表达水平越高,患者无进展生存期越短,预后越差(HR>1)(表2)。其中,1+2期卵巢癌中HR值为2.23,表明越早期的卵巢癌患者,MTERF1的预后价值越大。将截尾数据设置为OS,MTERF1的表达水平与卵巢癌患者的预后无显著相关性(P>0.05)。因此,早期(1+2期)卵巢癌患者中MTERF1mRNA水平越高,无进展生存期越短,预后越差(P<0.05)。
3 讨论

图3 10例卵巢癌与其相应的正常卵巢上皮组织中MTERF1蛋白表达水平的比值(*P<0.05)
卵巢癌在女性常见恶性肿瘤中占2.4%~6.5%,在女性生殖系统癌瘤中占第3位,次于宫颈癌和宫体癌。但在女性生殖系统癌瘤中,卵巢癌的病死率最高[13],75%的卵巢癌患者发现时已为晚期,并且具有比较明显的个体差异性[14]。目前,临床用于卵巢癌诊断的肿瘤标记物十分有限。发现能用于诊断和预后的卵巢癌分子标记物具有现实意义。
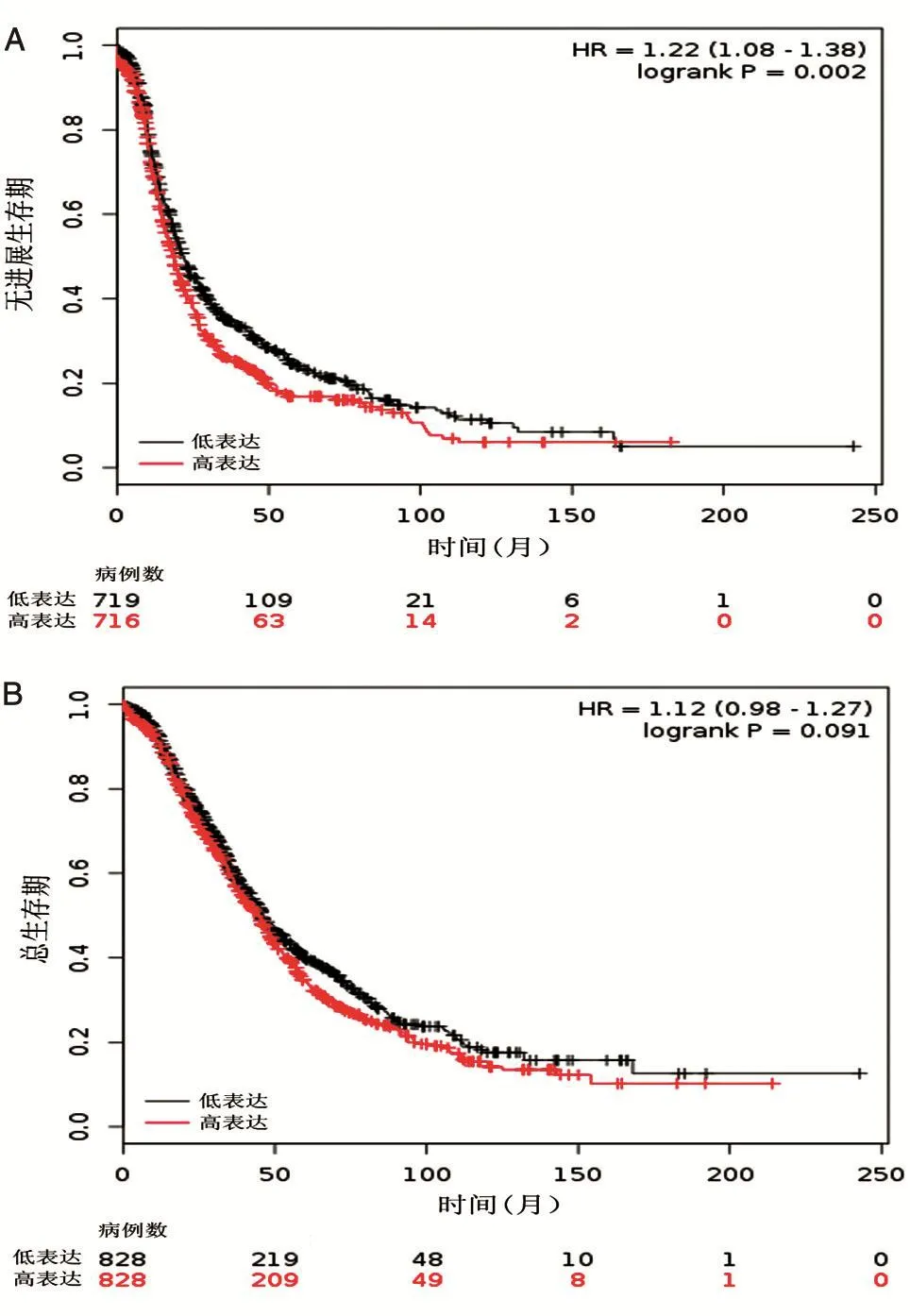
图4 MTERF1 mRNA表达量与卵巢癌患者预后的关系A:截尾数据为PFS;B:截尾数据为OS
MTERF1基因是MTERF超基因家族中最早被发现的成员。研究表明,与MTERF1蛋白结合的线粒体tRNALeu(UUR)基因A3243G位点的碱基突变,将引起线粒体糖尿病、线粒体脑肌病伴高?乳酸血症和卒中样发作综合征、进行性眼外肌麻痹等疾病。mtDNA靶序列A3243G点突变引起人MTERF1与DNA的亲合力下降,但并不改变线粒体转录产物的的比例,但线粒体蛋白质合成能力和细胞的呼吸活性下降将导致线粒体疾病[15]。目前,关于MTERF1基因在卵巢癌中的表达及其意义尚未见相关报道。Oncomine是一个大型肿瘤基因芯片分析整合数据库,提供公开、免费的整合分析资源,数据来源为公开发表的各种高通量测序数据。Oncomine平台为不熟悉高通量分析的科研工作者提供了获取整合信息的便捷途径,输入基因名称即可得到该基因在各种类型肿瘤中的表达差异等分析结果,并且以直观的形式输出[16]。通过对Oncomine数据库的整合分析,我们发现MTERF1在卵巢癌中显著高表达。我们分别采用荧光定量PCR和Western印迹对10例卵巢癌组织及其配对的正常卵巢上皮组织中MTERF1mRNA和蛋白质表达水平进行分析,发现6例卵巢癌组织中MTERF1高表达,其结果得到进一步证实。本研究的不足之处为样本数量有限,亟待扩大样本量进一步研究。
Kaplan-Meier plotter是目前被广泛接受的一个预后相关的在线分析数据库,涵盖了10 461例肿瘤样本,其中包括1816例卵巢癌样本、5143例乳腺癌样本、1065例胃癌样本和2437例肺癌样本,可以对54 675个基因进行相关预后分析,并得出真实可信的客观结果[17-18]。本研究首次通过Kaplan-Meier plotter数据库探讨MTERF1在卵巢癌及不同分期卵巢癌中的预后价值。结果显示,卵巢癌分期未明时,MTERF1表达水平越高,患者的无进展生存期越短,预后越差。但MTERF1表达水平与卵巢癌患者的总生存期无相关性。在1+2期卵巢癌,当截尾数据设置为PFS时,HR值为2.23,Log rankP<0.01;而当截尾数据为OS时,HR值为1.53,Log rankP=0.59。考虑到OS为总生存率,患者死亡原因可能存在非肿瘤致死,推测MTERF1可以较好地用于1+2期卵巢癌患者预后;而在晚期(3期及以后)卵巢癌患者中,无论是PFS还是OS,都与MTERF1的表达水平没有显著相关性,因此推测MTERF1对晚期卵巢癌不具备预后价值。

表2 各个分期卵巢癌患者的预后与MTERF1 mRNA水平的关系
综上所述,我们通过Oncomine和Kaplan-Mei⁃er plotter数据库对肿瘤相关基因的信息进行深入挖掘,提出MTERF1在卵巢癌组织中高表达;通过real time RT-PCR和Western印迹对卵巢癌及其配对的正常卵巢上皮组织中MTERF1mRNA和蛋白水平进行检测,证实MTERF1在卵巢癌组织中高表达;进而分析MTERF1表达对卵巢癌患者预后的影响,发现MTERF1与早期卵巢癌的预后有关,为进一步探讨MTERF1在卵巢癌发生发展中的作用提供了重要的理论依据和实验基础。
[1] Siegel R L,Miller K D,Jemal A.Cancer statistics, 2015[J].CA Cancer J Clin,2015,65(1):5-29.
[2] 李莉,熊国平,颜琳,等.CCNB1在卵巢癌中的表达及意义[J].现代妇产科进展,2016,25(11):818-820.
[3] 熊伟,杨勇琴,张海洋,等.人线粒体转录终止因子1 (hMTERF1)蛋白的生物信息学分析[J].生物信息学, 2015,13(1):23-30.
[4] 熊伟,余敏,左绍远.线粒体转录终止因子蛋白家族在线粒体基因表达中的调节作用[J].中国生物化学与分子生物学报,2015,31(3):223-231.
[5] Chen G,Dai J,Tan S,et al.MTERF1 regulates the oxidative phosphrylation activity and cell proliferation in C-33A cells[J].Acta Biochim Biophys Sin,2014, 46(6):512-521.
[6] 王巍,张志常,宋晓雯,等.基于数据挖掘分析PDCD5表达对胃癌预后的影响[J].现代肿瘤医学,2016,24(24): 3957-3959.
[7] Adib T R,Henderson S,Perrett C,et al.Predicting biomarkers for ovarian cancer using gene-expression microarrays[J].Br J Cancer,2004,90(3):686-692.
[8] Bonome T,Levine D A,Randonovich M,et al.A gene signature predicting for survival in subopimally debulked patients with ovarian cancer[J].Cancer Res, 2008,68(13):5478-5486.
[9] HendrixN D,Wu R,Kuick R,etal.Fibroblast growth factor 9 has oncogenic activity and is a down⁃stream target of Wnt signaling in ovarian endometri⁃oid adenocarcinomas[J].Cancer Res,2006,66(3):1354-1362.
[10]Lu K H,Patterson A P,Wang L,et al.Selection of potentialmarkersforepithelialovarian cancerwith gene expression arrays and recursive descent partition analysis[J].Clin Cancer Res,2004,10(10):3291-3300.
[11]Welsh J B,Zarrinkar P P,Sapinoso L M,et al.Anal⁃ysis of gene expression profiles in normal and neoplas⁃tic ovarian tissue samples identifies candidate molecu⁃lar markers of epithelial ovarian cancer[J].Proc Natl Acad Sci USA,2001,98(3):1176-1181.
[12]Yoshihara K,Tajima A,Komata D,et al.Gene expres⁃sion profiling of advanced-stage serous ovarian can⁃cers distinguishes novel subclasses and implicates ZEB2 in tumor pregression and prognosis[J].Cancer Sci,2009,100(8):1421-1428.
[13]宫艳秋,韩凤娟,吴效科,等.卵巢上皮性癌病因学研究进展[J].医学研究杂志,2010,39(5):18-20.
[14]朱秀娴,章晓乐,傅永伦,等.孕激素抑制LH峰在控制性卵巢刺激过程中的疗效观察[J].生殖与避孕,2015, 35(6):384-388.
[15]阳娅玲,肖红利,管敏鑫.人类线粒体tRNA生物合成与线粒体疾病[J].中国生物化学与分子生物学报, 2013,29(10):916-925.
[16]王进峰,卢晓明,王礼平,等.基于Oncomine芯片数据库荟萃分析碳酸苷酶IX在肾细胞癌中的表达及意义[J].现代泌尿生殖肿瘤杂志,2015,7(4):231-235.
[17]Szasz A M,Lanczky A,Nagy A,et al.Cross-valida⁃tion of survival associated biomarkers in gastric can⁃cer using transcriptomic data of 1065 patients[J].Onco⁃target,2016,7(31):49322-49333.
[18]Ocana A,Perez-Pena J,Alcaraz-Sanabria A,et al.In silico analyses identify gene-sets,associated with clini⁃cal outcome in ovarian cancer:role of mitotic kinases [J].Oncotarget,2016,7(16):22865-22872.
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年11期)2021-11-30
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中国生殖健康(2020年8期)2021-01-18
中国生殖健康(2018年1期)2018-11-06
安徽医科大学学报(2016年12期)2017-01-15
肿瘤预防与治疗(2015年2期)2015-09-26
医学研究杂志(2015年4期)2015-06-10
