
重组炭疽疫苗免疫人体产生抗体基因的生物信息学分析
2018-04-09 03:37:16刘渝娇陈郑珊王美荣张冠英范鹏飞房婷迟象阳于长明
生物技术通讯 2018年2期
刘渝娇,陈郑珊,王美荣,张冠英,范鹏飞,房婷,迟象阳,于长明
军事科学院 军事医学研究院 生物工程研究所,北京 100071
抗体是获得性免疫的重要组成部分,在体液免疫反应中起关键作用。其中单克隆抗体药物因具有高度靶向特异性、亲和力和安全性,已经在许多重大疾病的治疗、诊断和预防中得到广泛应用,是当今新药研发的重点。抗体基因序列的测定和分析是一个高速发展的科学领域,已经应用于多种疾病的诊断和防治中,包括疾病微小残留病变检测、移植的预后检测、疫苗系统性评价、中和抗体鉴定及B细胞运输模式追踪等[1-4]。随着高通量测序技术的发展和成熟,大规模、系统性分析抗体基因序列已成为可能。但是,抗体基因的胚系细胞多样性和体细胞多样性增加了分析的难度,特别是针对大容量、多样性高的抗体基因序列库,需要借助相应的生物信息学手段以及特定的计算方法对抗体基因序列进行分析和数据挖掘。
炭疽芽胞杆菌是一种致死率极高的人畜共患烈性传染病原。炭疽疫苗和抗体药物作为重要的炭疽防治手段,其研究一直是世界范围内关注的重点。本实验室研发的基因工程重组炭疽保护性抗原(protective antigen,PA)疫苗能够有效激发人体产生中和抗体。我们前期通过流式细胞分选-单细胞PCR技术,从重组PA疫苗免疫志愿者体内分选并扩增出大量重链和轻链天然配对的全人源单克隆抗体基因,包括34株PA结合抗体基因和4株中和抗体基因。在本研究中,我们在Linux操作系统(Ubuntu)上,基于NumPy、SciPy、pandas、Biopython、presto、Change-O[5]等构建了抗体基因序列分析平台,并利用alakazam、shazam、tigger等R语言包提供的函数进行编程实现相应功能,利用R、Perl等语言编写了一系列数据处理和接口程序,对前期获得的炭疽抗体可变区序列进行了一系列高效的生物信息学分析,包括抗体重链的VDJ基因使用和统计特征分析、基于聚类分析的抗体谱系树分析、中和抗体和结合抗体的重链CDR3区物理化学特性统计和分析、5-mer motifs体细胞高频突变模型构建。研究结果表明,前期获得的抗体基因序列中IGHV3、IGHD3和 IGHJ4的使用频率最高,IGHV3和IGHD5、IGHD6和IGHJ4、IGHV3和IGHJ4的组合是序列中出现频率最高的组合方式;23.3%的PA结合抗体都出现在同一个抗体聚类(239号克隆组)中;中和抗体CDR3区较其他抗体对碱性和酸性氨基酸的使用频率较低,偏向于使用疏水性氨基酸;抗体的体细胞高频突变多发生在AAAAC、GTTAA、AGCTC、AAGTA等热点motif。另外,运用ARGalaxy Web Sever[6-7]的抗体基因序列分析模块得到相应的数据可视化结果。
本研究搭建的抗体基因序列分析平台整合安装了完整的序列处理和分析程序,可离线使用。该平台实现了对高通量抗体基因序列的快速处理分析,而且具有较好的拓展性,可自由组合使用各个功能模块以及编程开发满足实际需求的新功能。本研究结果对于重组炭疽疫苗评价和生物信息学辅助的抗体鉴定和改造具有一定的借鉴意义,提供的方法也可为其他相关疫苗激发免疫系统应答产生抗体谱的研究及中和抗体的筛选和鉴定提供帮助。
1 材料与方法
1.1 抗体基因序列来源
采集重组PA疫苗免疫志愿者的血液样品,利用密度梯度离心法分离外周血单个核细胞(PBMC),进行荧光染色,通过流式细胞分选仪从PBMC中分选单个抗体分泌细胞克隆(CD19+/ CD3-/CD20-/CD27high/CD38high)。采用反转录PCR从单个B细胞中扩增抗体基因,再以该PCR产物为模板进行单细胞巢式PCR反应,经琼脂糖凝胶电泳鉴定为阳性的巢式PCR产物送上海生工生物工程股份有限公司测序,批量整理测序结果即得到抗体可变区基因序列,保存为单个fasta格式文件。共计346条H链可变区序列(其中4条序列为有中和活性的抗体,30条序列为仅有结合活性的抗体)、268条κ链可变区序列、173条λ链可变区序列。考虑到抗体的特性主要由H链可变区决定,故未对κ和λ链可变区基因序列进行类似分析。
1.2 基因序列的注释和标准数据集的生成
由于使用反向测序引物,因而需要将测序所得的抗体基因序列进行反向互补后再进行后续分析。利用BioEdit[8]软件批量操作,得到全部原始基因序列的反向互补序列。利用平台中本地化的IgBLAST程序对抗体可变区基因序列进行注释,IgBLAST程序具有识别序列的FR/CDR边界、最佳匹配胚系V(D)J基因、显示V(D)J基因连接细节、提供核苷酸的重排连接信息等功能。进而运用Change-O程序集提供的MakeDb预处理程序(Python语言编写)对IgBLAST程序的输出文件进行整理和格式转化,生成可供平台内其他应用程序使用的标准tab-delimited数据集文件。
1.3 基于抗体基因标准数据集的分析方法
对得到的标准数据集文件在平台内进行处理分析,使用R语言包alakazam[10]提供的count⁃Gene函数对抗体重链基因数据集进行VDJ基因使用情况的统计,可得到VDJ基因节段数量、节段家族数量等统计信息。R语言包shazam[5]提供的distToNearest函数用于计算调整抗体基因序列分层聚类的合适阈值[11],并将该阈值作为Defines⁃Clone程序(由Change-O程序集提供)的dist参数值。进而运行DefineClones以定义克隆组(clone group),对抗体基因序列进行聚类。使用Perl语言编写的程序对已聚类的序列进行克隆组选择和数据处理,使用PHYLIP[12]软件包中的dnapars程序为同一克隆组内的抗体序列建立谱系树,并通过R语言包igraph调整和绘制谱系树网络图。al⁃akazam包提供的countClones函数可用于统计克隆组的情况。alakazam包中还提供了一系列用于分析抗体氨基酸序列理化性质的函数,如利用ami⁃noAcidProperties函数分析序列CDR3区的长度、亲疏水性和氨基酸的酸碱性等特征。shazam包提供的createTargetingModel函数[13]可通过序列信息构建出体细胞高频突变模式模型(SHM targeting models),并通过plotMutability函数将模型可视化为直观的猬形图(Hedgehog)。在ARGalaxy Web Sever上传序列文件后选择ARGalaxy模块下Im⁃mune Repertoire pipeline功能,根据实际情况选择参数,提交运行后得到分析结果。另外,Galaxian Web Sever的可视化模块提供Circos Builder功能,提交相关数据文件和配置文件可在线绘制Circos图[14]。
2 结果
2.1 抗体基因序列分析平台和流程
将测序结果用BioEidt软件处理得到反向互补序列,在搭建的平台上对反向互补序列文件进行一系列数据处理和序列分析。具体流程为,用本地IgBLAST程序注释后再用MakeDb.py进行格式转换,生成标准tab-delimited数据集文件,用countGene函数进行VDJ基因使用统计分析、De⁃fineClones程序进行抗体序列聚类分析、dnapars程序绘制抗体谱系树、createTargetingModel函数构建体细胞高频突变模式模型、aminoAcidProperties函数分析CDR3区氨基酸理化性质,完成序列分析。图1显示了所搭建的抗体基因序列分析平台和抗体基因序列分析的流程。
2.2 V(D)J基因使用
通过R语言包alakazam中的countGene函数统计抗体重链VDJ基因使用情况(图2),包括节段基因家族、节段基因和等位基因的统计结果。结果显示VDJ基因使用频率最高的胚系基因节段家族分别为IGHV3、IGHD3、IGHJ4,使用频率最高的胚系基因分别为IGHV3-23、IGHD3-10,使用频率最高的等位基因分别为IGHV3-23*01、IGHD6-13*01、IGHJ4*02。
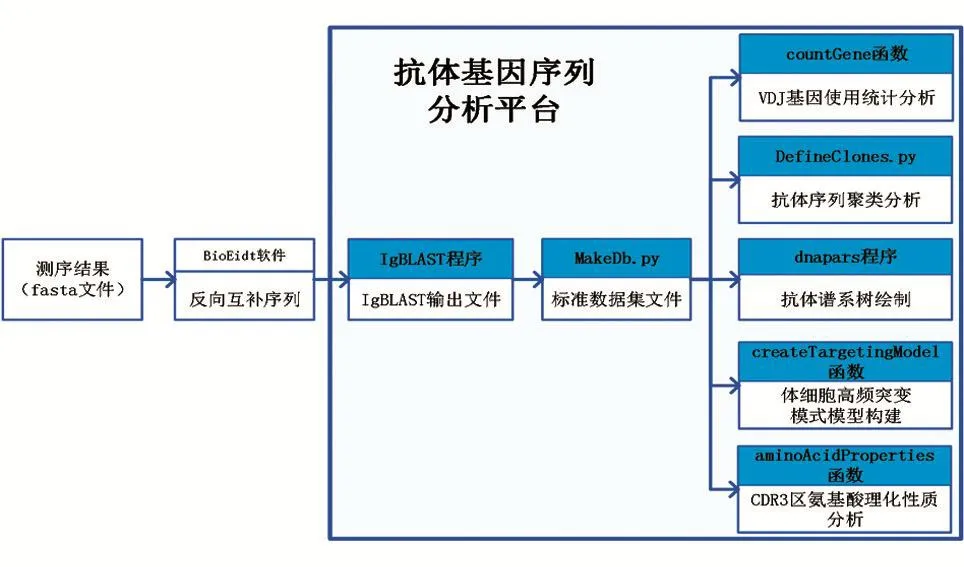
图1 抗体基因序列分析流程

图2 VDJ基因的部分统计结果A:VDJ基因家族使用统计;B:VDJ基因使用统计;C:VDJ等位基因使用统计
另外,ARGalaxy Web Sever生成的VDJ基因使用情况的柱状统计图(图3)显示使用频率最高的胚系基因节段家族分别为IGHV3、IGHD3、IGHJ4,使用频率最高的胚系基因分别为IGHV3-23、IGHD3-10,与平台分析得到的结果一致。
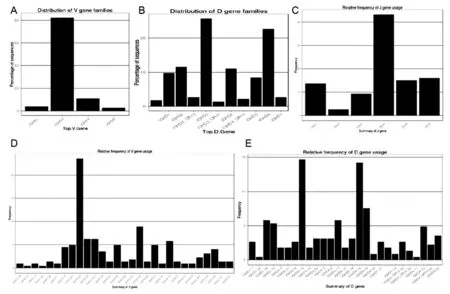
图3 VDJ基因使用频率统计图A:V基因家族使用统计;B:D基因家族使用统计;C:J基因家族使用统计;D:V基因使用统计;E:D基因使用统计
ARGalaxy Web Sever生成的Circos图和Heat⁃map图(图4)反映抗体重链VDJ基因的组合情况。图4A、D表现D基因和J基因的组合,B、E表现V基因和D基因的组合,C、F表现V基因和J基因的组合。Circos图圆周上的每段弧代表特定基因,2段弧之间的连线(连接条带)即表示2个基因的组合情况,连接条带越宽则表明在统计结果中该组合情况出现的频率越高。Heatmap图的横轴和纵轴分别表示2类节段基因,图中的方格点表征特定基因的组合情况,方格点的颜色越深(偏蓝色)表明在统计结果中该组合出现的频率越高。从Circos和Heatmap图可以看出,DJ基因组合出现频率较高的是IGHD6-13和IGHJ4、IGHD-10和IGHJ4、IGHD3-10和IGHJ6;VD基因组合出现频率较高的是IGHV3-64和IGHD6-13、IGHV3-23和IGHD4-24、IGHV3-23和IGHD3-10;VJ基因组合出现频率较高的是IGHV3-23和IGHJ4、IGHV3-64和IGHJ4、IGHV3-23和IGHJ6。

图4 VDJ基因组合情况A:D基因和J基因的组合统计Circos图;B:V基因和D基因的组合统计Circos图;C:D基因和J基因的组合统计Circos图;D:D基因和J基因的组合统计Heatmap图;E:V基因和D基因的组合统计Heatmap图;F:D基因和J基因的组合统计Heatmap图
2.3 Clone聚类分析
2.3.1 克隆组的定义 基于抗体基因序列间的汉明距离(Hamming distance)对序列进行聚类,定义克隆组,将每条序列都归入特定的“克隆组”。根据该模型的定义,“克隆组”内的抗体序列可能源自相同的B细胞。利用DefineClones程序对所研究的序列数据集划分了254个克隆组,同一克隆组内的序列数目从1条至26条不等。定义只包含1条序列的克隆组为单序列克隆组,则有中和活性抗体的4条重链序列均分布在单序列克隆组中;仅有结合活性抗体的总共30条重链序列中,22条分布在单序列克隆组中,7条集中分布在239号克隆组中,1条分布在36号克隆组中。图5显示了克隆组内序列数量最多的前15个克隆组。
2.3.2 生成B细胞谱系树(lineage trees) PHY⁃LIP软件包中的dnapars程序为239号“克隆组”内的抗体基因序列生成抗体谱系树(图6),结合抗体基因所在的节点位置用红圈标记,根节点是胚系基因。dnapars程序基于最大简约法(maximum parsimony)建树,该算法常用于系统发生学分析,根据离散型性状(如基因序列)的变异程度,构建系统发育树和分析演化关系。
2.4 重链CDR3区物理化学性特性分析
考虑到抗体重链的CDR3区在抗原抗体相互作用中的重要性,统计分析了其氨基酸序列的理化特性,包括CDR3区长度、氨基酸使用情况等。图7显示中和抗体CDR3区较其他抗体对碱性和酸性氨基酸的使用频率较低,偏向于使用疏水性氨基酸。
2.5 体细胞高频突变模型

图5 包含序列数量最多的15个克隆组

图6 第239号“克隆组”的抗体谱系树
针对给定的抗体基因序列数据集,构建出5个碱基长度motif模式的体细胞高频突变模型(SHM targeting models),反映特定突变发生的可能性。模型结果以猬形图的形式表现,猬形图主要由5层圆环和外围放射状线条构成,第3层圆环表示特定的碱基(A、T、G、C中的某一种),内外侧4个圆环上依次循环排布着ATGC等4种碱基(颜色区别)。因此,A图表示NNANN模式motifs,B图表示NNTNN模式motifs,C图表示NNCNN模式motifs,D图表示NNGNN模式motifs,其中N表示4种碱基中的某一种。沿固定半径从内圈到外圈即可读取到特定的5个碱基长度的motif,其对应的放射状线条的长度表示发生突变的可能性。放射状线条的不同颜色分别表示传统的hotspots(热点)和cold-spots(冷点),hot-spots标记为红色(WRC/GYW)和绿色(WA/TW),cold-spots标记为蓝色(SYC/GRS)。从图8可以看出,抗体的体细胞发生高频突变更可能发生在AAAAC、GT⁃TAA、AGCTC、AAGTA等热点motif。
3 讨论
前期通过采集重组PA疫苗免疫志愿者血液样品,并进行流式细胞分选和单细胞PCR快速扩增出全人源单克隆抗体基因,对于测序后得到大量的抗体可变区基因序列,采用简单的序列比对和基础的统计方法已经远远不能满足批量序列分析和生物学信息提取的需要。本研究采取了一系列行之有效的生物信息学手段处理和研究序列信息,取得较好的分析结果,这些流程和方法也同样适用于更高通量的抗体基因序列的处理和分析。
抗体重链可变区基因包括V基因片段(vari⁃able segment)、D基因片段(diversity segment)和J基因片段(joining segment)。对VDJ基因使用情况的统计显示,对于IgHV基因,使用频率最高的胚系基因节段家族为IgHV3,其次是IgHV4。仅发现IgHV1、IgHV3、IgHV4、IgHV5等4个胚系基因节段家族的使用,这可能与单细胞PCR引物的偏性或抗体序列有限的样本量有关。对于IgHD基因,使用频率最高的胚系基因节段家族为IgHD3。对于IgHJ基因,使用频率最高的胚系基?因节段家族为IgHJ4。这些结果与预期及以前的报道相符[15-16]。VDJ基因组合方式的频率也有较大差别,IgHV3-23和IgHJ4、IgHV3-64和IgHD6-13、IgHD6-13和IgHJ4组合出现在同一条抗体重链基因上的可能性最大。可视化的统计结果同时直观地表明了抗体基因的组合多样性,VDJ基因在B细胞发育过程中发生重排,从而形成多种多样的组合形式,产生丰富的多样性。轻链基因结构与此类似,只是缺少D基因片段。

图7 重链CDR3区氨基酸序列理化特性
对克隆组的定义和序列的聚类分析发现,23.3%的PA结合抗体都出现在同一个抗体聚类(239号克隆组)中,而具有中和活性的重链可变区基因序列更倾向于分布在单序列克隆组(克隆组内只有一条序列样本),即中和抗体的重链基因序列比较“独特”,与数据集中的其他序列具有演化关系的可能性较小。同时,中和抗体CDR3区较其他抗体对碱性和酸性氨基酸的使用频率较低,偏向于使用疏水性氨基酸,在炭疽中和抗体亲和力成熟改造和中和活性提高的过程中,可以参考上述结果,对可变区的改造朝着特定聚类方向,或者使用疏水性氨基酸从而提高抗体的亲和力和中和活性。

图8 SHM模型猬形图A:NNANN模式motifs;B:NNTNN模式motifs;C:NNCNN模式motifs;D:NNGNN模式motifs
体细胞高频突变也是抗体多样性的重要来源,其在抗原刺激后作用于已重排过的V基因上。其实质是抗体基因(尤其是在突变热点位置,主要是CDR区)发生的点突变,而且发生频率很高。突变后有些分子的亲和力会优于原先的分子,从而发生亲和力成熟的现象。在统计抗体基因序列数据集信息基础上,构建出体细胞高频突变模式模型,模型可视化表现为猬形图的形式,直观反映5个碱基长度的motif的突变可能性以及hot-spots和cold-spots的分布情况,对于抗体改造靶点的确定有一定的指导意义。如果能使用更高通量的抗体组库序列信息,并且做对照分析(如比较免疫前后的抗体基因变化,比较不同物种之间抗体基因的差异等),则构建的细胞高频突变模式模型更具科学性和实际意义。本研究结果提供了一种将DNA和蛋白质的功能、结构和进化信息联系起来的生物信息学手段,可为其他疾病的相关研究提供参考。
[1]Robins H.Immunosequencing:applications of immune repertoiredeep sequencing[J].CurrOpin Immunol, 2013,25(5):646-652.
[2] Stern J N,Yaari G,Vander Heiden J A,et al.B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes[J].Sci Transl Med, 2014,6(248):248ra107.
[3] Khurana S,Fuentes S,Coyle E M,et al.Human anti⁃body repertoire after VSV-Ebola vaccination identifies noveltargets and virus-neutralizing IgM antibodies [J].Nat Med,2016,22(12):1439-1447.
[4] Wang B,Kluwe C A,Lungu O I,et al.Facile discov⁃ery of a diverse panel of anti-Ebola virus antibodies by immune repertoire mining[J].SciRep,2015,5: 13926.
[5] Gupta N T, Heiden J A V, Uduman M, et al. Change-O:a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data[J].Bioinfor⁃matics,2015,31(20):3356.
[6] Ijspeert H,van Schouwenburg P A,Van Z D,et al. Antigen receptor galaxy:a user-friendly,Web-based tool for analysis and visualization of T and B cell re⁃ceptor repertoire data[J].J Immunol,2017,198(10):415 6-4165.
[7] Moorhouse M J.ImmunoGlobulin galaxy(IGGalaxy)for simple determination and quantitation of immunoglobu⁃lin heavy chain rearrangements from NGS[J].BMC Im⁃munol,2014,15(1):59.
[8] Hall T A.BioEdit:a user-friendly biological sequence alignment editor and analysis program for Windows 95/ 98/NT[J].Nucleic Acids Symp Ser,1999,41(41):95-98.
[9] Ye J,Ma N,Madden T L,et al.IgBLAST:an immu⁃noglobulin variabledomain sequenceanalysistool[J]. Nucleic Acids Res,2013,41(Web Server issue):W34.
[10]Stern J N,Yaari G,Vander Heiden J A,et al.B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes[J].Sci Transl Med, 2014,6(248):248ra107.
[11]Glanville J,Zhai W,Berka J,et al.Precise determina⁃tion of the diversity of a combinatorial antibody li⁃brary givesinsightinto the human immunoglobulin repertoire[J].Proc Natl Acad Sci USA,2009,106(48): 20216-20221.
[12]Felsenstein J.PHYLIP-phylogenetic inference package (Version 3.2)[J].Cladistics,1989,5(4):164-166.
[13]Gur Y,Vander H J A,Mohamed U,et al.Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immu⁃noglobulin sequencing data[J].Front Immunol,2013,4: 358.
[14]Krzywinski M,Schein J,Birol I,et al.Circos:an in⁃formation aesthetic forcomparative genomics[J].Ge⁃nome Res,2009,19(9):1639-1645.
[15]Laserson U,Vigneault F,Gadalamaria D,et al.Highresolution antibody dynamics of vaccine-induced im⁃mune responses[J].Proc Natl Acad Sci USA,2014,111 (13):4928-4933.
[16]Mroczek E S,Ippolito G C,Rogosch T,et al.Differ⁃ences in the composition of the human antibody reper⁃toire by B Cell subsets in the blood[J].Front Immu⁃nol,2014,5(5):96.
猜你喜欢
中国港湾建设(2022年12期)2022-12-28 05:27:24
环球时报(2022-09-20)2022-09-20 15:18:57
今日农业(2020年24期)2020-12-15 16:16:00
中国比较医学杂志(2018年6期)2018-06-26 07:27:22
科教导刊(2017年10期)2017-06-09 18:38:05
心血管病防治知识(2016年9期)2016-11-21 02:45:24
兽医导刊(2016年12期)2016-05-17 03:51:50
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:58
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:58
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
