
非CAG三核苷酸异常扩增相关脊髓小脑共济失调患者CACNA1A基因突变分析
2018-04-02 09:39杨志华刘玉涛骆海洋唐秘博王燕琳史长河许予明
郑州大学学报(医学版) 2018年2期
张 槊,杨志华,刘玉涛,骆海洋,唐秘博,杨 靖,王燕琳,史长河,许予明
郑州大学第一附属医院神经内科 郑州 450052
脊髓小脑性共济失调(spinocerebellar ataxia, SCA)是一组具有临床及遗传异质性的神经退行性疾病[1],主要累及小脑及相关神经传入传出通路,临床上以小脑性共济失调为主要特征,伴有锥体系及锥体外系疾病症状[2]。其遗传方式以常染色体显性遗传为主,目前已有至少44个基因或位点被确定与SCA的发病相关(http://neuromuscular.wustl.edu/ataxia/domatax.html)。CAG三核苷酸重复所致的SCA亚型,如SCA1,2,3,6,7,17和齿状核红核苍白球路易体萎缩症(DRPLA)占所有SCA病例的50%以上,但是其他一些常规的基因所导致的SCA则较少报道[1]。与CAG异常扩增所致的SCA相比,其他基因所引起的共济失调多表现为疾病进展缓慢,生存期长,致残率低等特点[1]。CACNA1A(NM_000068.3)编码神经元电压依赖性的P/Q型钙通道α-1亚基(Cav2.1),该亚基参与调控钙离子调控进入细胞并在多种钙离子调控的生命活动中有着重要作用,比如基因表达、神经递质释放等[3]。CACNA1A在小脑及大脑中广泛表达[4]。既往研究报道CACNA1A变异参与SCA6型(SCA6)[5]、发作性共济失调2型(episodic ataxia type 2, EA2)[6]、家族性偏瘫型偏头痛(familial hemiplegic migraine, FHM)[7]的致病机制。SCA6是由CACNA1A的第1号外显子中编码谷氨酰胺的三核苷酸CAG过度扩增所致异常蛋白引起[5],FHM和EA2则主要是由该基因的点突变所致[8-11]。近年来研究[12]报道CACNA1A除参与以上3种疾病以外,还可以通过该基因的点突变等引起SCA。Coutelier 等[13]报道CACNA1A是SCA中除包含CAG重复各基因以外的最常见的致病基因。为了解CACNA1A基因在中国SCA患者中的突变情况,本研究应用PCR结合DNA序列分析方法,对初步排除包含CAG重复基因的68例SCA家系先证者进行了CACNA1A基因的突变分析,现将结果报道如下。
1 对象与方法
1.1研究对象郑州大学第一附属医院神经内科2010年6月至2016年6月所收集的68例SCA家系的先证者,均已排除CAG重复扩增的亚型SCA1,2,3,6,7,17和DRPLA。由两名神经内科医师进行详细的病史采集、神经系统查体,详细询问家族史,完善影像学等检查。同时收集该院2015年1月至2016年6月200名无神经精神系统疾病的健康志愿者为对照。研究得到郑州大学第一附属医院伦理委员会批准,并取得受试者的知情同意。
1.2基因测序及产物分析取患者及健康志愿者外周静脉血5~10 mL,抗凝,抽提基因组DNA。根据NCBI网站CACNA1A外显子序列,应用Primer 6设计引物(表1),由苏州金唯智生物科技有限公司合成。PCR反应体系为30 μL:DNA模板2 μL,正、反向引物(20 μmol/L)各1 μL,2×Taq PCR Master Mix(南京诺唯赞生物科技有限公司)15 μL,超纯水11 μL。反应条件:95 ℃预变性10 min;95 ℃变性1 min,应用touchdown的方式退火1 min,72 ℃延伸 1 min,共35个循环;最后72 ℃延伸10 min,4 ℃存放。取10 μL反应产物在20 g/L的琼脂糖凝胶中电泳,之后进行goldview染色并在紫外线下观察结果。将上述PCR产物送至华大基因直接测序。测序结果应用Chromas软件分析,并与人类基因组基因(GenBank序列号 NC_000019.10)比对。将筛选到的可能致病位点在200名健康志愿者中依照前述PCR扩增及测序方法进行验证。

表1 CACNA1A引物列表

续表1
2 结果
2.1CACNA1A基因的测序结果对68例SCA家系先证者进行测序检测后,在1例先证者(图1)中检出CACNA1A基因第19号外显子2 992位到2 997位碱基发生GAGGGC缺失变异,该突变导致其所编码蛋白Cav2.1第998谷氨酸和999位甘氨酸发生缺失(图2)。在200名健康志愿者中未发现该突变位点。
2.2临床资料及辅助检查结果先证者(Ⅱ-3),女,33岁,以“走路不稳”为主诉入院。4 a前无明显诱因出现走路不稳,宽基底步态,步速较前减慢,伴言语不利,构音障碍,后出现间断性双手及头部不自主抖动,运动或静止时均可出现,发病以来,记忆力略有减退。神经系统查体示:右上肢肌力5级,左上肢肌力近端5级、远端4级,双下肢肌力近端4级、远端4级;四肢肌张力正常,深浅感觉未见异常;深反射正常,四肢病理征阴性。头部MRI示小脑萎缩(图3)。该家系中先证者的父亲(Ⅰ-1)患有同样疾病,于33岁发病,临床表现为走路不稳,双手及头部不自主抖动。但由于先证者的父亲于43岁时去世,我们未能获得更多的临床资料。
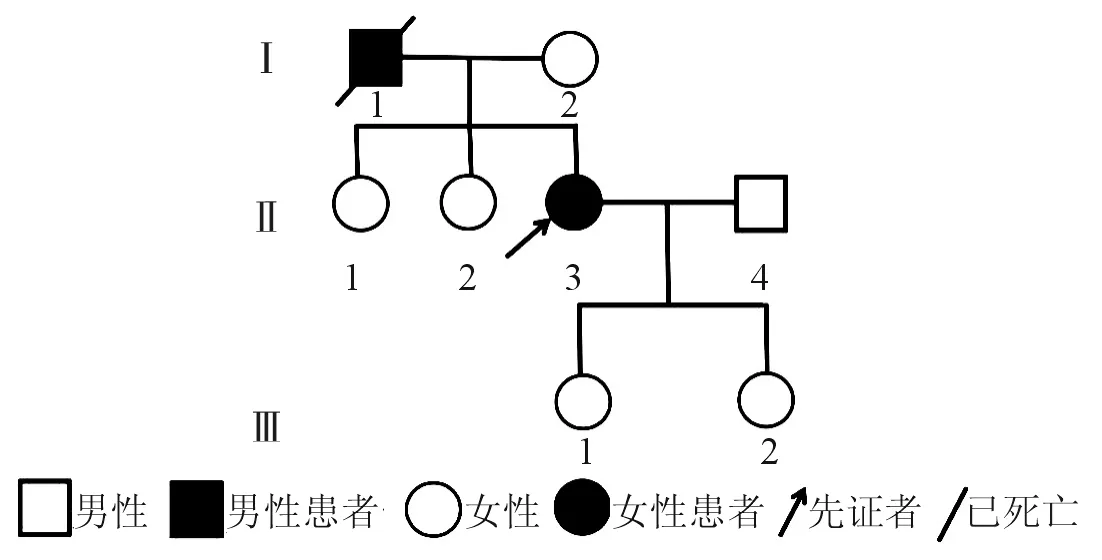
图1 家系系谱图

图2 CACNA1A突变测序(箭头所示为碱基缺失位点)
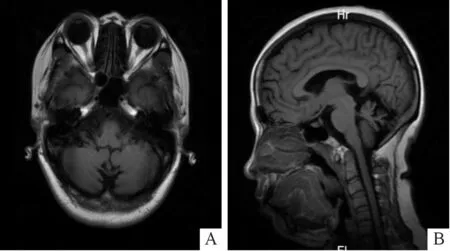
A:轴位,B:矢状位图3 头颅MRI
3 讨论
CACNA1A基因位于19号染色体,编码神经元电压依赖性的P/Q型钙离子通道α-1亚基CaV2.1。CaV2.1作为一种电压门控钙离子通道,广泛分布于大脑、海马、小脑和脊髓神经元的突触前膜和树突[14-16],并调节神经元突触囊泡的释放[17-19]。在小脑中,CaV2.1门控通道参与调控蒲肯野细胞和颗粒细胞的钙离子流及细胞活性[20-21]。此外,小脑蒲肯野细胞或颗粒细胞中CACNA1A的敲除可以导致蒲肯野细胞的活性下降进而引起小脑功能的改变[22-23]。
这些研究表明CACNA1A在调节蒲肯野细胞活性及小脑功能中发挥着重要作用。
近年来CACNA1A基因除了CAG重复次数异常扩增以外,其他变异如点突变等导致SCA亦有报道[12,24-25]。中国曾有研究[26]报道CACNA1A第25号外显子c.G4034GA (p.R1345Q)点突变可以导致共济失调并伴有震颤。此外,另有研究[13]报道CACNA1A基因点突变等变异是SCA中除CAG重复异常扩增基因如ataxin 1、ataxin 2、ataxin 3等以外出现频率最高的致病原因。这些证据表明CACNA1A在SCA的发病中有着重要作用,在中国SCA患者中进行CACNA1A基因的检测显得很有必要。
本研究在来自中国的SCA家系的1例先证者中发现了一个CACNA1A基因的缺失型变异,c.2992-2997del(p.Glu998-Gly999del)。该患者表现为缓慢进展的走路不稳,构音障碍,并伴有手和头部的不自主抖动,同时影像学表现为小脑萎缩。其临床表现区别于SCA6患者,SCA6患者除共济失调以外,多伴随有严重的吞咽障碍、复视、锥体束征等;同时也区别于EA2,该病多表现为间歇发作的共济失调,尤其是在疾病的初始阶段[6]。一系列研究报道CACNA1A突变可以导致共济失调:S218L突变可以导致共济失调并表现有癫痫症状[27-28];R1352Q突变则表现为早发型共济失调并伴有偏头痛[12]。故可以认为CACNA1A变异在SCA发病中有着重要的作用,虽然不同位点的突变所导致的症状有很大差异。这些不同可能是因为突变所导致的蛋白构象改变或是相关遗传因子的影响。所以,CACNA1A基因序列的测定将进一步完善SCA的基因诊断,尤其是排除CAG异常扩增之后。
研究[3]表明,CACNA1A的变异可导致CaV2.1蛋白的稳定性和离子通道的活性下降。本研究中的CACNA1A基因的缺失变异导致CaV2.1蛋白第998位谷氨酸和999位甘氨酸的缺失,其可能导致CaV2.1生理功能的损伤。碱基缺失所致的非框移突变引起共济失调的情况在其他基因中也有报道,如一个日本的SCA5家系就是由SPTBN2基因的c.2608_2610delGAG缺失所致,该缺失导致所编码蛋白第870位谷氨酸发生缺失[29]。但是,CACNA1A的缺失型变异导致共济失调的致病机制仍需要进一步的探索。
综上所述,我们在68例来自SCA家系的先证者中发现了1例CACNA1A基因第19号外显子第2 992位到2 997位碱基发生GAGGGC缺失变异,引起Cav2.1第998位谷氨酸和999位甘氨酸发生缺失。尽管CACNA1A基因突变比例较小,但该研究结果丰富了SCA致病基因数据库,提示进行SCA基因筛查时勿遗漏CACNA1A基因检测。
[1] DURR A.Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond[J].Lancet Neurol,2010,9(9):885
[2] JAYADEV S,BIRD TD.Hereditary ataxias:overview[J].Genet Med,2013,15(9):673
[3] PIETROBON D.CaV2.1 channelopathies[J].Pflugers Arch,2010,460(2):375
[4] PIETROBON D,STRIESSNIG J.Neurobiology of migraine[J].Nat Rev Neurosci,2003,4(5):386
[5] ZHUCHENKO O,BAILEY J,BONNEN P,et al.Autosomal dominant cerebellar ataxia(SCA6)associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel[J].Nat Genet,1997,15(1):62
[6] JODICE C, MANTUANO E, VENEZIANO L, et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6(SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p[J]. Hum Mol Genet, 1997,6(11):1973
[7] HAAN J,KORS EE,VANMOLKOT KR,et al.Migraine genetics:an update[J].Curr Pain Headache Rep,2005,9(3):213
[8] HANS M,LUVISETTO S,WILLIAMS ME,et al.Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine[J].J Neurosci,1999,19(5):1610
[9] JEN JC,GRAVES TD,HESS EJ,et al.Primary episodic ataxias: diagnosis, pathogenesis and treatment[J].Brain,2007,130(Pt 10):2484
[10]RAJAKULENDRAN S,GRAVES TD,LABRUM RW,et al.Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy[J].J Physiol,2010,588(Pt11):1905
[11]TOTTENE A,FELLIN T,PAGNUTTI S,et al.Familial hemiplegic migraine mutations increase Ca(2+)influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons[J].Proc Natl Acad Sci U S A,2002,99(20):13284
[12]TRAVAGLINI L,NARDELLA M,BELLACCHIO E,et al.Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia[J].Eur J Paediatr Neurol,2017,21(3):450
[13]COUTELIER M,COARELLI G,MONIN ML,et al.A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies[J].Brain,2017,140(6):1579
[14]ROSSIGNOL E,KRUGLIKOV I,VAN DEN MAAGDENBERG AM,et al.CaV 2.1 ablation in cortical interneurons selectively impairs fast-spiking basket cells and causes generalized seizures[J].Ann Neurol,2013,74(2):209
[15]LUDWIG A,FLOCKERZI V,HOFMANN F.Regional expression and cellular localization of the alpha1 and beta subunit of high voltage-activated calcium channels in rat brain[J].J Neurosci,1997,17(4):1339
[16]WESTENBROEK RE,SAKURAI T,ELLIOTT EM,et al.Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels[J].J Neurosci,1995,15(10):6403
[17]ZAITSEV AV,POVYSHEVA NV,LEWIS DA,et al.P/Q-type,but not N-type,calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex[J].J Neurophysiol,2007,97(5):3567
[18]CAO YQ,TSIEN RW.Effects of familial hemiplegic migraine type 1 mutations on neuronal P/Q-type Ca2+ channel activity and inhibitory synaptic transmission[J].Proc Natl Acad Sci U S A,2005,102(7):2590
[19]ALI AB,NELSON C.Distinct Ca2+channels mediate transmitter release at excitatory synapses displaying different dynamic properties in rat neocortex[J].Cereb Cortex,2006,16(3):386
[20]JUN K,PIEDRAS-RENTERLA ES,SMITH SM,et al.Ablation of P/Q-type Ca(2+) channel currents,altered synaptic transmission,and progressive ataxia in mice lacking the alpha(1A)-subunit[J].Proc Natl Acad Sci U S A,1999,96(26):15245
[21]LAU FC,ABBOTT LC,RHYU IJ,et al.Expression of calcium channel alpha1A mRNA and protein in the leaner mouse(tgla/tgla)cerebellum[J].Brain Res Mol Brain Res,1998,59(1):93
[22]MAEJIMA T,WOLLENWEBER P,TEUSNER LU,et al.Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice[J].J Neurosci,2013,33(12):5162
[23]MARK MD,MAEJIMA T,KUCKELSBERG D,et al.Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations[J].J Neurosci,2011,31(11):4311
[24]BURK K,KAISER FJ,TENNSTEDT S,et al.A novel missense mutation in CACNA1A evaluated by in silico protein modeling is associated with non-episodic spinocerebellar ataxia with slow progression[J].Eur J Med Genet,2014,57(5):207
[25]NAIK S,POHL K,MALIK M,et al.Early-onset cerebellar atrophy associated with mutation in the CACNA1A gene[J].Pediatr Neurol,2011,45(5):328
[26]JIANG HS,WANG DM,WANG Q,et al. Missense mutation R1345Q in CACNA1A gene causes a new type of ataxia with episodic tremor: clinical features, genetic analysis and treatment in a familial case[J].Nan Fang Yi Ke Da Xue Xue Bao,2016,36(7):883
[27]TOTTENE A,PIVOTTO F,FELLIN T,et al.Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma[J].J Biol Chem,2005,280(18):17678
[28]STAM AH,LUIJCKX GJ,POLL-THE BT,et al.Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation[J].J Neurol Neurosurg Psychiatry,2009,80(10):1125
[29]WANG Y, KOH K, MIWA M, et al. A Japanese SCA5 family with a novel three-nucleotide in-frame deletion mutation in the SPTBN2 gene: a clinical and genetic study[J]. J Hum Genet, 2014,59(10):569
猜你喜欢
中国医药导报(2022年28期)2022-11-25
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
中国医学影像技术(2020年11期)2020-01-13
创新作文(小学版)(2019年4期)2019-07-24
郑州大学学报(医学版)(2019年3期)2019-06-03
中南林业科技大学学报(2019年4期)2019-04-08
