
载气对ZrO2-Al2O3催化正丁烷异构化反应性能的影响
2018-03-22 16:16:11张文芳张敏秀王鹏照杨朝合李春义
石油炼制与化工 2018年7期
张文芳,张敏秀,王鹏照,杨朝合,李春义
(中国石油大学(华东)重质油国家重点实验室,山东 青岛 266580)
碳四资源的深度利用是目前亟需解决的难题。碳四资源的主要组成有正丁烷、异丁烷、正丁烯、异丁烯和丁二烯,碳四原料经丁二烯抽提和甲基叔丁基醚(MTBE)工艺处理后,还剩余大量的正丁烷和异丁烷。其中,正丁烷用途少,而异丁烷用途广泛。异丁烷与丁烯烷基化生产烷基化油、与丙烯共氧化制备环氧丙烷并联产叔丁醇或MTBE[1]。正丁烷异构化,可将低附加值的正丁烷转变为高附加值的异丁烷,为解决正丁烷过剩和异丁烷可能出现的供不应求问题提供了一条切实可行的技术路线[2]。
1 实 验
1.1 原 料
ZrOCl2·8H2O、Al(NO3)3·9H2O、氨水、硫酸,均为分析纯,国药集团化学试剂有限公司生产;正丁烷纯度大于99.995%;氢气纯度99.999%;氮气纯度99.999%。
1.2 催化剂的制备

1.3 催化剂反应性能评价
催化剂的异构化反应性能评价在固定床微型反应评价装置上进行。催化剂装填量为1.0 g,以正丁烷为原料,氢气、氮气及其混合气为载气。具体操作条件如下:将催化剂在空气氛围(30 mLmin)下活化30 min,活化温度450 ℃;然后在温度200 ℃、常压条件下,保持气体线速不变,通过改变载气与原料的摩尔比,考察载气气氛对SZA催化剂上正丁烷异构化反应性能的影响。原料和产物组成由Bruker GC450气相色谱仪分析。
正丁烷转化率=(1-气相产物中正丁烷质量分数)×100%
正丁烷转化率降低幅度(xh)=[正丁烷初始转化率(0 h)-正丁烷转化率(xh)]正丁烷初始转化率(0 h)×100%;x为反应时间。
2 结果与讨论
2.1 n(H2)∶n(C4+H2)对正丁烷异构化反应的影响
在保持气体线速不变的前提下,选取n(H2)∶n(C4+H2)分别为0,0.2,0.4,0.6,0.8,考察n(H2)∶n(C4+H2)对SZA催化剂催化正丁烷异构化反应性能的影响。
2.1.1n(H2)∶n(C4+H2)对正丁烷转化率的影响不同n(H2)∶n(C4+H2)条件下,SZA催化剂作用下的正丁烷异构化反应结果见图1。从图1(a)可以看出:随着n(H2)∶n(C4+H2)的升高,正丁烷的转化率逐渐增加,当n(H2)∶n(C4+H2)低于0.4时,正丁烷转化率缓慢上升,而当n(H2)∶n(C4+H2)升高至大于0.6时,正丁烷转化率显著提高,表明氢气的加入对正丁烷转化具有促进作用;随着反应时间的延长,正丁烷转化率不断降低,催化剂逐渐失活,n(H2)∶n(C4+H2)对催化剂的失活行为影响显著;n(H2)∶n(C4+H2)小于0.4时,初始条件下正丁烷转化率迅速降低,反应1 h后催化剂失活速率逐渐减小;在n(H2)∶n(C4+H2)大于等于0.6的条件下,随反应时间的延长,正丁烷转化率呈平缓降低趋势,当n(H2)∶n(C4+H2)达到0.8时,反应5 h后,正丁烷转化率从54.08%下降至40.71%,转化率降低幅度仍高达24.7%(图1(b))。从中可以得出:一方面,氢气的引入可以起到减缓催化剂失活的作用,且n(H2)∶n(C4+H2)越高,催化剂失活速率越慢(图1(b));另一方面,氢气的引入对提高催化剂稳定性的能力有限,即使在很高的n(H2)∶n(C4+H2)条件下,催化剂仍不可避免地发生失活现象。

n(H2)∶n(C4+H2):■—0; ●—0.2; ▲—0.4; ◆—0.8

■—(X0h-X5h)X0h; ▲—(X1h-X5h)X1h图1 n(H2)∶n(C4+H2)对正丁烷异构化反应的影响
上述实验证实氢气在反应过程中是非惰性的,即氢气不仅仅起到稀释反应物和中间产物的作用,还与SZA催化剂发生相互作用,从而对异构化反应性能产生影响。①正丁烷在SZA催化剂上发生异构化反应时,单双分子反应机理同时存在[11-12],而双分子反应涉及到丁烯和丁基正碳离子二聚生成C8中间物种。向反应体系中引入氢气后,氢气与正丁烷分子在活性中心上发生竞争吸附,且随着n(H2)∶n(C4+H2)的升高,丁烯和丁基碳正离子在催化剂床层的浓度降低,发生二聚反应和与C8中间物种发生缩合生焦反应的几率也越低,从而提高了催化剂的稳定性[13]。②S=O双键强吸电子作用促进了与硫酸物种相连的Zr—O键对氢气的吸附作用,使氢气在其表面发生异裂形成高活性的氢供体,促进不饱和中间物种发生加氢饱和反应,实现从催化剂表面酸性位上的快速脱附(见图2)。这有效抑制了缩合生焦等副反应的发生,从而显著提高催化剂的稳定性[8]。Domen等[14]通过原位红外技术在3 668 cm-1和1 562 cm-1处分别检测到了由氢异裂生成的O—H 和Zr—H键,也证实了这一观点。
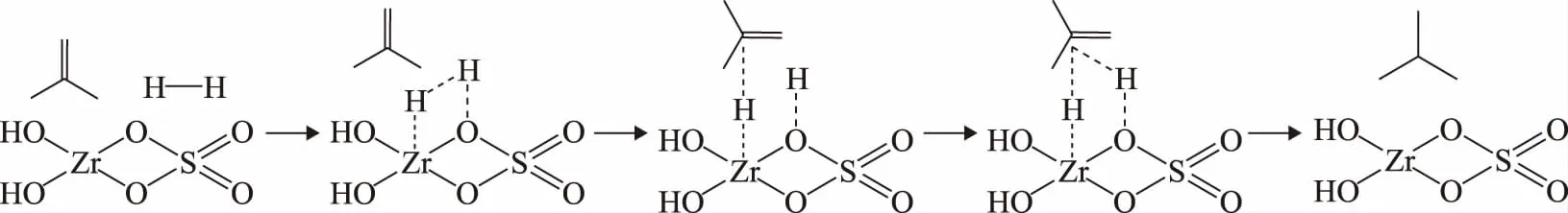
图2 SZA催化剂上氢气分解和异丁烷加氢的路径示意
上述有关氢气对催化剂稳定性的影响也可以用来解释n(H2)∶n(C4+H2)对正丁烷初始转化率的影响。不同n(H2)∶n(C4+H2)条件下,正丁烷与新鲜催化剂接触瞬间表现出相同的异构化活性。考虑到本实验中将反应前10 min的平均结果作为初始反应性能,稳定性的提高在宏观上表现反应前10 min正丁烷转化率的增加。
2.1.2n(H2)∶n(C4+H2)对异丁烷选择性的影响图3为不同n(H2)∶n(C4+H2)条件下异丁烷选择性随反应时间的变化。从图3可以看出:随着n(H2)∶n(C4+H2)由0增加到0.8,异丁烷初始选择性由89.89%上升至92.09%,即增加n(H2)∶n(C4+H2)有利于提高异丁烷的初始选择性;随着反应时间的延长,异丁烷选择性呈上升趋势;反应前10 min正丁烷选择性在90%左右。反应1 h后,异丁烷选择性升高至94%左右。
图3 不同n(N2)∶n(C4+N2)对异丁烷选择性的影响n(H2)∶n(C4+H2):■—0; ●—0.2; ▲—0.4; ◆—0.8
2.2 n(N2)∶n(C4+N2)对正丁烷异构化反应的影响

n(N2)∶n(C4+N2):■—0; ●—0.2; ▲—0.4; ◆—0.8

■—(X0h-X5h)X0h; ▲—(X1h-X5h)X1h图4 不同n(N2):n(C4+N2)对正丁烷异构化反应的影响
考察n(N2)∶n(C4+N2)对SZA催化剂活性的影响,结果见图4。从图4(a)可以看出:随着n(N2)∶n(C4+N2)的增加,正丁烷转化率也逐渐增加,表明氮气的加入对正丁烷转化也有促进作用。加入不同比例的氮气正丁烷转化率增加的趋势大致相同,与加入氢气实验现象不同的是,未出现n(N2)∶n(C4+N2)达到一定值,转化率显著提高的现象;随着反应时间的延长,正丁烷转化率不断下降,催化剂逐渐失活。与n(H2)∶n(C4+H2)条件下的实验现象不同的是,n(N2)∶n(C4+N2)对催化剂的失活行为基本没有影响,不同n(N2)∶n(C4+N2)条件下转化率的降低趋势基本一致(图4(b)),即催化剂失活行为一致。
上述实验表明,与氢气相比,氮气在反应过程中是惰性的,即氮气仅仅起到稀释反应物和中间产物在催化剂床层浓度的作用,与催化剂没有相互作用。
2.3 氮气、氢气混合气中不同n(H2)∶n(N2)对正丁烷异构化反应的影响
从2.1节可以得知,氢气对异构化反应的影响有两个方面:一是稀释反应物和中间产物在催化剂床层的浓度;二是与催化剂发生相互作用。但究竟哪方面是主要原因,哪是次要原因,还是两者起到的作用相等,对此,难以给出定论。从2.2节得知,氮气仅仅起到稀释的作用,由此,设计出本节实验,即保持总压不变(稀释效果不变),改变n(H2)∶n(N2)(改变了氢气与催化剂间相互作用的强弱),考察不同n(H2)∶n(N2)对正丁烷异构化反应的影响,结果见图5。由图5(a)可以看出,该实验条件下正丁烷转化率变化趋势与不同n(H2)∶n(C4+H2)条件下基本相同,两者初始转化率和转化率降低幅度变化趋势(图5(b))均一致。因此,稀释效果所起的作用不大,催化剂稳定性的提高是由氢气与催化剂的相互作用实现的。
3 结 论

n(H2)∶n(N2)∶n(C4):■—0.6∶0∶0.4; ●—0.45∶0.15∶0.4; ▲—0.3∶0.3∶0.4; ◆—0∶0.6∶0.4

■—(X0h-X5h)X0h; ▲—(X1h-X5h)X1h图5 不同n(H2)∶n(N2)对正丁烷异构化反应的影响
SZA催化正丁烷异构化反应过程中,载气的类型和摩尔比对催化剂反应行为具有显著影响。氮气作为载气是惰性的,仅仅起到稀释原料和中间产物浓度的作用;氢气是非惰性的,除稀释作用外,还与催化剂发生相互作用,一方面与正丁烷分子在活性中心上发生竞争吸附,降低不饱和中间物种在催化剂表面酸性位上的浓度,另一方面还能提高中间物种和产物分子从催化剂表面上脱附的能力,有效抑制了缩合生焦等副反应的发生,从而减缓了催化剂的失活速率。其中,与催化剂的相互作用是提高SZA催化剂稳定性的关键因素。
参考文献
[1] 李涛.碳四馏分中异丁烷的利用方案研究[J].石油化工技术与经济,2015(2):1-6
[4] 张艺菲.固体超强酸异构化催化剂的失活与再生[D].上海:华东理工大学,2012
[5] 张六一,韩彩芸,杜东泉,等.硫酸化氧化锆固体超强酸[J].化学进展,2011,23(5):860-873

[7] Villegas J I,Kumar N,Heikkilä T,et al.A highly stable and selective Pt-modified mordenite catalyst for the skeletal isomerization ofn-butane[J].Applied Catalysis A:General,2005,284(12):223-230
[8] Hong Z,Fogash K B,Dumesic J A.Reaction kinetic behavior of sulfated-zirconia catalysts for butane isomerization[J].Catalysis Today,1999,51(2):269-288
[9] Wang Pengzhao,Zhang Jiaoyu,Wang Guowei,et al.Nature of active sites and deactivation mechanism forn-butane isomerization over alumina-promoted sulfated zirconia[J].Journal of Catalysis,2016,338:124-134
[11] 邹艳.正丁烷异构化的反应机理和催剂制备研究[D].上海:复旦大学,2006
[12] Nieminen V,Kangas M,Salmi T,et al.Kinetic study ofn-butane isomerization over Pt-H-mordenite[J].Industrial & Engineering Chemistry Research,2005,44(3):471-484
[13] Villegas J I,Kumar N,Heikkilä T,et al.Isomerization ofn-butane to isobutane over Pt-modified Beta and ZSM-5 zeolite catalysts:Catalyst deactivation and regeneration[J].Chemical Engineering Journal,2006,120(1):83-89
[14] Kondo J,Domen K,Maruya K,et al.Infrared study of molecularly adsorbed H2on ZrO2[J].Chemical Physics Letters,1992,188(5):443-445
猜你喜欢
炼油与化工(2021年3期)2021-07-06 11:12:52
化工管理(2020年19期)2020-07-28 03:05:34
石油石化绿色低碳(2019年6期)2019-01-14 01:16:20
石油石化绿色低碳(2019年6期)2019-01-14 01:16:12
合成化学(2015年10期)2016-01-17 08:56:29
化工进展(2015年3期)2015-11-11 09:09:44
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:13
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:13
化学反应工程与工艺(2015年3期)2015-04-16 03:06:19
化学工业与工程(2015年1期)2015-02-10 03:01:34
