
高效液相色谱法测定盐酸黄酮哌酯原料和制剂中有关物质
2018-03-20 05:49沈丹丹梁涵雁姜学美
中国药业 2018年5期
沈丹丹 ,曾 杰 ,梁涵雁 ,姜学美 ,蒋 晨
(1.重庆市食品药品检验检测研究院,重庆 401121; 2.重庆市药物过程与质量控制工程技术研究中心,重庆401121; 3.福安药业<集团>股份有限公司,重庆 401121)
盐酸黄酮哌酯为第2代尿失禁治疗药物,具有抑制腺苷酸环化酶、磷酸二酯酶及拮抗钙离子作用,并有弱抗毒蕈碱作用,对泌尿生殖系统的平滑肌具有选择性解痉作用,临床主要用于治疗各种尿路疾病引起的尿频、尿急、尿痛、排尿困难及尿失禁等症[1-7]。国内盐酸黄酮哌酯原料有6家企业生产,但现在市场供应的原料只有2家企业生产。目前,国内盐酸黄酮哌酯有片剂和胶囊剂,有 0.1 g和 0.2 g 2个规格,其中片剂有 10家生产企业,胶囊剂有2家生产企业。本次国家药品抽样评价共抽取样品107批,全部为片剂,涉及生产企业6家,未抽到胶囊剂。
盐酸黄酮哌酯原料、片剂和胶囊剂的现行质量标准均为2015年版《中国药典(二部)》[8],其有关物质检查方法均为薄层色谱(TLC)法,曾有报道采用高效液相色谱(HPLC)法测定盐酸黄酮哌酯含量[9],但目前国内尚无盐酸黄酮哌酯有关物质测定方法的研究报道。国内上市的盐酸黄酮哌酯原料和制剂有关物质执行标准均为现行药典标准,采用薄层色谱(TLC)法。该方法为半量法,操作比较烦琐,方法灵敏度较低,人为判断较主观,且仅控制杂质3-甲基黄酮-8-羧酸。107批样品按照现行质量标准检验,仅有30批样品检出已知杂质A(3-甲基黄酮-8-羧酸),仅1批样品检出未知杂质,说明现行药典有关物质测定方法专属性和灵敏度均较差,检查结果不能反映药品中杂质的真实含量。美国药典(USP39)[10]、英国药典(BP2016)[11]收载的盐酸黄酮哌酯原料有关物质检查方法均为HPLC法,但色谱条件和杂质限度不相同;盐酸黄酮哌酯片有关物质USP39采用HPLC法,BP2016为TLC法。USP39收载的原料杂质谱包括3-甲基黄酮-8-羧酸(杂质A)、3-甲基黄酮-8-羧酸甲酯(杂质 B)、3-甲基黄酮-8-羧酸乙酯(杂质C),BP2016收载的原料杂质谱包括3-甲基黄酮-8-羧酸(杂质A)、3-甲基黄酮-8-羧酸乙酯(杂质B)、3-甲基黄酮-8-羧酸异丙酯(杂质C),因此考虑结合USP39及BP2016的杂质谱。本研究中建立了同时测定 3-甲基黄酮-8-羧酸(杂质 A)、3-甲基黄酮 -8-羧酸甲酯(杂质B)、3-甲基黄酮 -8-羧酸乙酯(杂质C)及3-甲基黄酮-8-羧酸异丙酯(杂质D)的HPLC法,并制订合理的质控指标,供现行药典质量标准提高参考。现报道如下。
1 仪器与试药
仪器:Waters e2695型高效液相色谱仪,包括Waters 2489型紫外检测器(美国Waters公司);K51502653型电子天平(德国赛多利斯公司)。
试药:盐酸黄酮哌酯对照品(中国食品药品检定研究院,批号为100964-200701,含量为100.0%);杂质A(3-甲基黄酮-8-羧酸)对照品(中国食品药品检定研究院,批号为100965-200701,含量为100.0%);杂质 B(3-甲基黄酮-8-羧酸甲酯)对照品(USP,批号为1.0,含量 100.0% );杂质 C(3- 甲基黄酮 -8- 羧酸乙酯)对照品(BP,批号为 156.02.08.01,含量为 99.9% );杂质 D(3-甲基黄酮 -8-羧酸异丙酯)对照品(BP,批号为2417,含量为99.4%)。盐酸黄酮哌酯片,A厂(批号为 20150602),B 厂(批号为 150504),C 厂(批号为20160304),D 厂 (批号为 20150501),E 厂 (批号为03A160207),F 厂(批号为 20150701),规格均为每片0.2 g;盐酸黄酮哌酯原料,F 厂(批号为 20160502),G厂(批号为FA160142)。己烷磺酸钠(白赛勤化学技术有限公司,批号为20161125),三乙胺(成都市科龙化工试剂厂,批号为20161011),磷酸(重庆川东化工有限公司,批号为20170601),均为分析纯;乙腈(安徽时联特种溶剂股份有限公司,批号为6308IS317)为色谱纯。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Phenomenex Luna C18柱(150 mm ×4.6 mm,5 μm),十八烷基硅烷键合硅胶为填充剂;流动相:缓冲液(取1 g己烷磺酸钠,加水适量使溶解,加三乙胺1 mL和磷酸 1 mL,加水稀释至 650 mL,摇匀)-乙腈(65∶35)等度洗脱1 min,待第1个色谱峰(盐酸黄酮哌酯峰)洗脱完毕后,线性梯度洗脱(见表1);检测波长:293 nm;流速:1.0 mL /min;进样量:20 μL。取混合对照品溶液20 μL注入液相色谱仪,先以流动相等度洗脱,待第1个色谱峰(盐酸黄酮哌酯峰)洗脱完毕,按表1进行线性梯度洗脱。盐酸黄酮哌酯、杂质A、杂质B、杂质 C、杂质D依次出峰,各峰间分离度均符合规定,色谱图见图1。

表1 流动相梯度洗脱程序
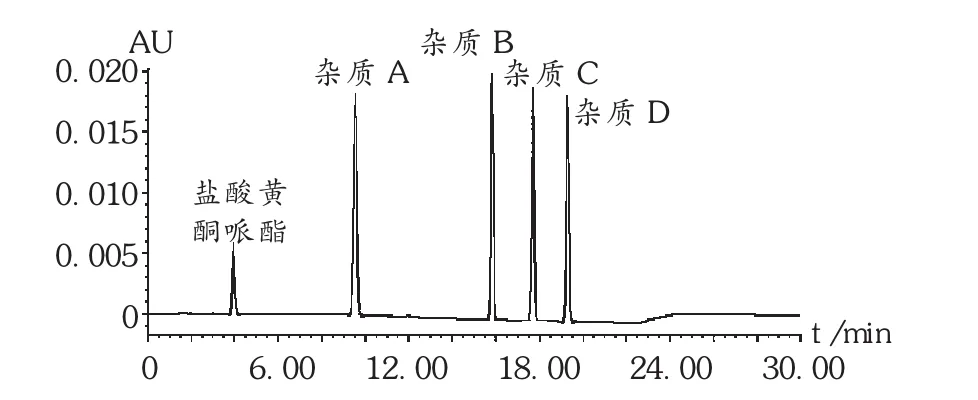
图1 系统适用性试验高效液相色谱图
2.2 溶液制备
取本品细粉适量(约相当于盐酸黄酮哌酯100 mg),置100 mL容量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。取盐酸黄酮哌酯对照品、杂质A对照品、杂质B对照品、杂质C对照品、杂质D对照品各约10 mg,精密称定,分别置100 mL容量瓶中,用溶剂稀释至刻度,摇匀,分别作为对照品贮备液;精密量取盐酸黄酮哌酯对照品贮备液1.0 mL,杂质 A、杂质 B、杂质C及杂质 D贮备液各3.0 mL,置100 mL容量瓶中,用溶剂稀释至刻度,作为混合对照品溶液。
2.3 专属性试验
按照各个厂家处方配制混合辅料,精密量取辅料适量,依法配制成辅料溶液,精密量取续滤液20 μL,注入液相色谱仪,记录色谱图。在此条件下,空白溶剂、空白辅料均不干扰有关物质的测试,方法专属性良好(见图 2)。
2.4 破坏性试验
光照破坏:取紫外光(254 nm)下照24 h的盐酸黄酮哌酯原料25 mg,精密称定,置25 mL容量瓶中,加溶剂使溶解,稀释至刻度,摇匀,即得供试品溶液。
氧化破坏:取盐酸黄酮哌酯原料25 mg,精密称定,置25 mL容量瓶中,加30%过氧化氢5 mL,放置5 min,加溶剂稀释至刻度,摇匀,即得供试品溶液。
酸破坏:取盐酸黄酮哌酯原料25 mg,精密称定,置25 mL 容量瓶中,加 1.0 mol/L 盐酸 1 mL,放置 10 min,分别加1.0 mol/L氢氧化钠1 mL,加溶剂稀释至刻度,摇匀,即得供试品溶液。
碱破坏:取盐酸黄酮哌酯原料25 mg,精密称定,置25 mL 容量瓶中,加 1.0 mol/L 氢氧化钠 1 mL,放置10 min,分别加 1.0 mol/L 盐酸 1 mL,加溶剂稀释至刻度,摇匀,即得供试品溶液。
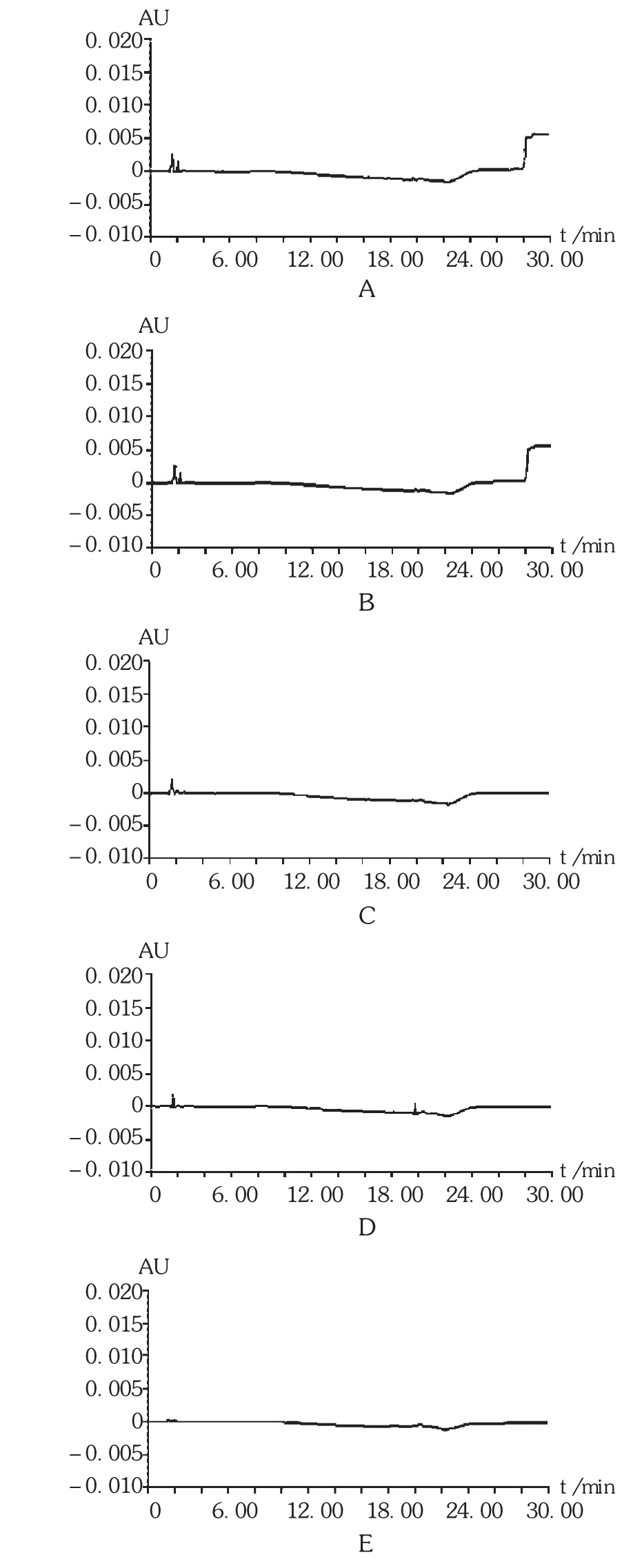

图2 专属性试验高效液相色谱图
高温破坏:取盐酸黄酮哌酯原料适量,分别置烘箱中60℃条件下放置24 h,取出放至室温,精密称定25 mg,置25 mL容量瓶中,用溶剂溶解并稀释至刻度,摇匀,即得供试品溶液。
高湿破坏:取盐酸黄酮哌酯原料适量,分别在温度为25℃、相对湿度为92.5%条件下放置10 d,取出,放至室温。取本品各约25 mg,置25 mL容量瓶中,用溶剂溶解并稀释至刻度,摇匀,即得供试品溶液。
未破坏:取盐酸黄酮哌酯原料适量,依法制备供试品溶液。
精密量取上述供试品续滤液20 μL,注入液相色谱仪,记录色谱图(见图3)。按峰面积归一化法计算杂质含量,结果见表2。与未破坏样品杂质含量比较,结果F厂和G厂家的原料对碱均不稳定。其中,F厂原料碱破坏条件下杂质A含量(58.60%)显著性增加,G厂原料在强碱条件下杂质A含量(2.10%)也增加,表明在强碱条件下容易降解产生杂质A。其他条件下杂质含量基本无变化,表明供试品溶液对光、高温、高湿、酸及氧均比较稳定。
2.5 线性关系考察
称取盐酸黄酮哌酯对照品10.81 mg,杂质A对照品 9.74 mg,杂质 B 对照品 10.21 mg,杂质 C 对照品10.30 mg,杂质 D 10.77 mg,精密称定,分别置 100 mL容量瓶中,用溶剂稀释至刻度,摇匀,分别作为对照品贮备液。精密量取盐酸黄酮哌酯对照品贮备液1.0 mL,杂质 A,B,C,D 贮备液各 0.30,0.75,1.50,3.00,4.50,6.00 mL,分别置100 mL容量瓶中,用溶剂稀释至刻度,作为混合对照品溶液。取混合对照品溶液各20 μL,分别注入液相色谱仪,记录色谱图。以质量浓度为横坐标、峰面积为纵坐标绘制标准曲线,得回归方程。结果见表3。
2.6 各杂质的校正因子计算
根据各杂质和盐酸黄酮哌酯线性试验中线性曲线斜率,计算各杂质与盐酸黄酮哌酯的曲线斜率之比即为校正因子。结果见表 3。可见,杂质 A,B,C,D校正因子均不在0.90~1.10范围内,故不能采用自身对照法测定杂质含量。
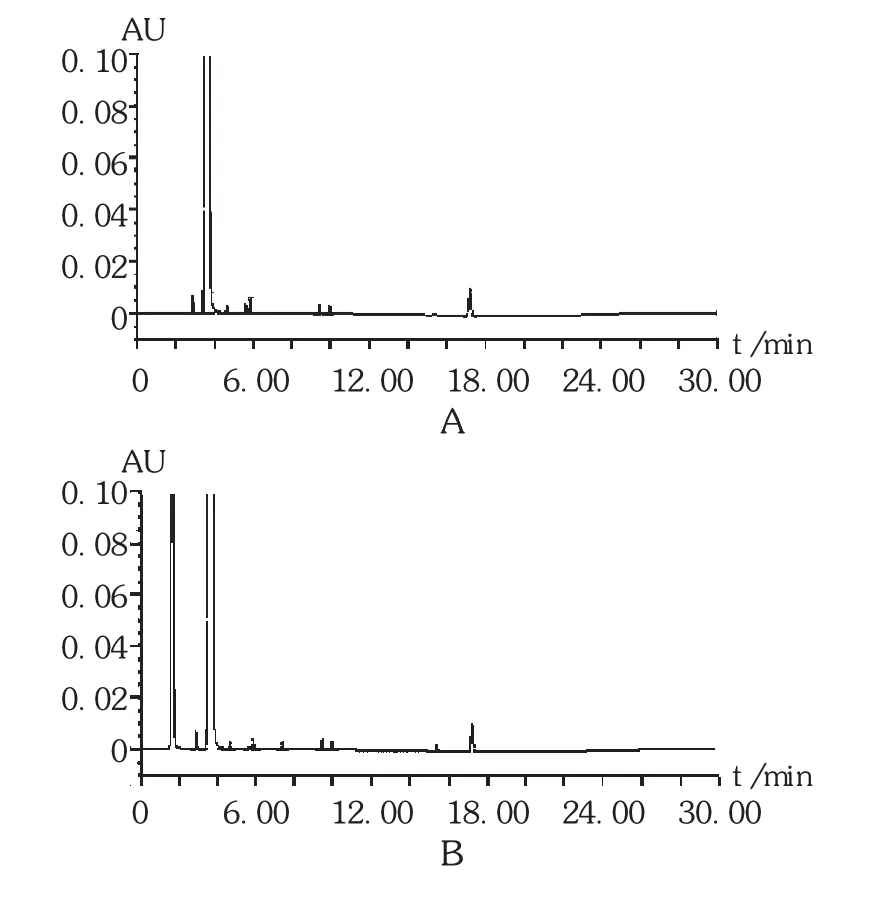
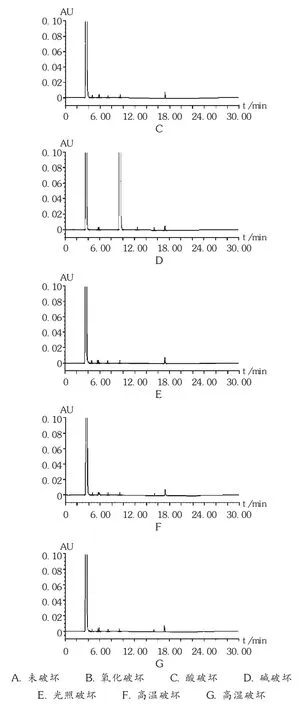
图3 破坏性试验高效液相色谱图
2.7 精密度试验
精密吸取混合对照品溶液20 μL,平行6次,注入液相色谱仪,记录色谱图。结果见表3。可见,仪器精密度良好。
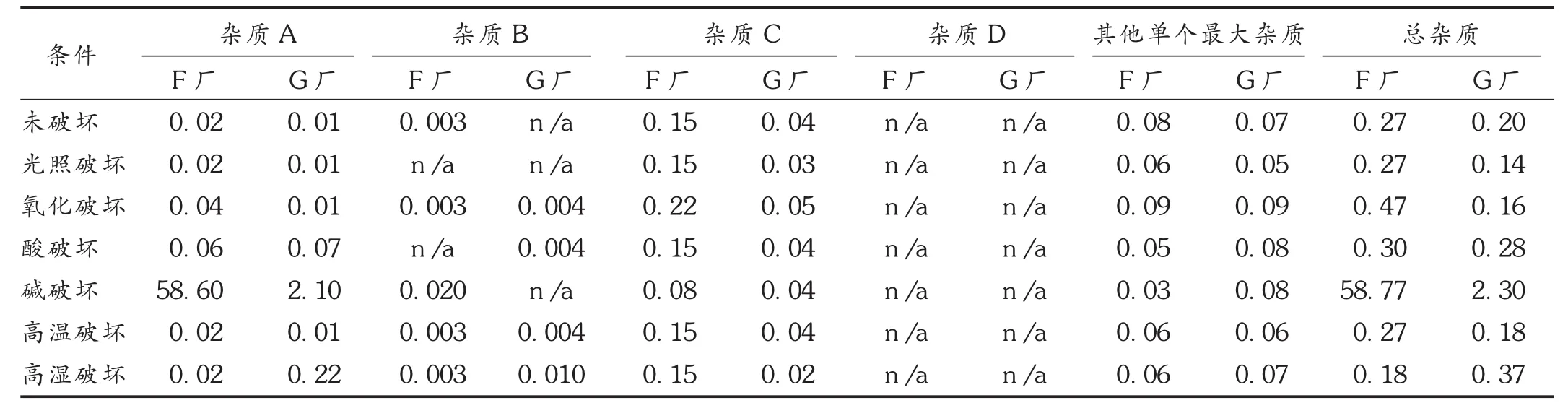
表2 F厂和G厂原料破坏性试验结果(%)
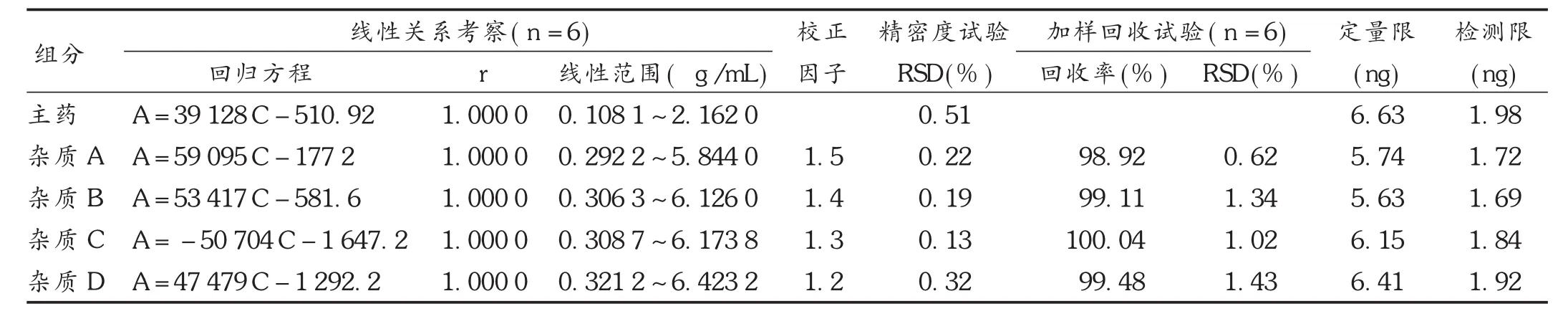
表3 方法学考察试验结果
2.8 加样回收试验
取本品内容物细粉适量(约相当于盐酸黄酮哌酯100 mg),精密称定,平行12份,分别置100 mL容量瓶中。分别精密加入杂质 A,B,C,D 质量浓度为 100 μg/mL的对照品溶液 0.3,3.0,6.0 mL,各 3 份,用流动相稀释至刻度,摇匀,使各杂质质量浓度约相当于供试品溶液测定质量浓度的 0.03% ,0.30% ,0.60% ;最后 2 份用流动相稀释至刻度,摇匀;将上述各溶液超声10 min,放冷,滤过,取续滤液作为供试品溶液。另精密量取杂质A,B,C,D对照品适量,用流动相溶解并稀释成每1 mL中含3 μg的混合溶液作为对照品溶液。精密量取供试品溶液与对照品溶液各20 μL,分别注入液相色谱仪,记录色谱图。按外标法分别计算每份供试品中各杂质的总量,计算回收率。结果见表3。
2.9 定量限与检测限测定
精密量取2.5项下混合对照品溶液适量,用溶剂逐级稀释成系列质量浓度,吸取20 μL注入液相色谱仪,记录色谱图。以信噪比(S/N)3∶1计算各成分的检测限,以信噪比(S/N)10∶1计算定量限。结果见表 3。
2.1 0 稳定性试验
取盐酸黄酮哌酯片(F厂,批号为20160102)适量,研细,按 2.2项下方法制备供试品溶液,分别于0,1,2,3,4,6,8,12,16,20 h 时吸取 20 μL 注入液相色谱仪,记录色谱图。计算各已知杂质、其他单个最大杂质、其他总杂质峰面积的 RSD,以评价溶液的稳定性。杂质A在20 h内峰面积的 RSD为2.9%,说明杂质A不稳定(在6 h内的 RSD为0.4%,说明杂质A在6 h内稳定);杂质B及杂质C在20 h内的 RSD分别为1.6%和0.9%,低于2.0%;杂质D未检出;其他单个最大杂质在20 h内的 RSD为0.6%;总杂质在6 h内的 RSD为0.2%,表明供试品溶液在6 h内稳定性好。
2.1 1 耐用性试验
色谱柱:色谱柱选用 Phenomenex Gemini C18柱(150 mm ×4.6 mm,5 μm),Phenomenex Luna C18柱(150mm ×4.6mm,5μm),资生堂Spolar C18柱(150mm ×4.6 mm,5 μm)及岛津 Inertsil ODS-C18柱(150 mm ×4.6 mm,5 μm),其他条件保持不变,精密量取 2.1 项下系统适用性溶液20 μL,注入液相色谱仪,记录色谱图。结果不同种类的色谱柱对分离度无明显影响,本方法对色谱柱的种类耐用性强。
柱温:将柱温分别设定为25,30,35℃,其他条件保持不变,精密量取 2.1项下系统适用性溶液 20 μL,注入液相色谱仪,记录色谱图。结果柱温在25~35℃变化范围内,分离度均大于2.0,表明方法对柱温耐用性强。
流速:将流速分别设定为 0.8,1.0,1.2 mL /min,其他条件保持不变,精密量取2.1项下系统适用性溶液20 μL,注入液相色谱仪,记录色谱图。结果流速在0.8~1.2 mL /min 变化范围内,分离度均大于 2.0,表明方法对流速耐用性强。
2.1 2 样品含量测定
取6个厂家样品,研细,按2.2项下方法制备供试品溶液,精密量取 20 μL,注入液相色谱仪,记录色谱图,按外标法计算杂质含量。结果见表4。
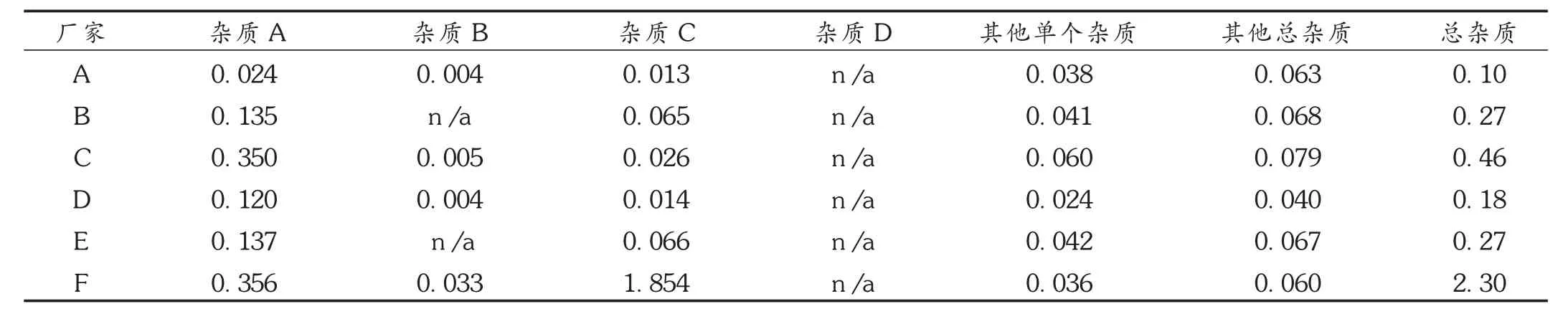
表4 各厂家样品有关物质测定结果(%)
3 讨论
3.1 色谱条件选择
盐酸黄酮哌酯极性较强,容易被洗脱,但杂质A,C,D极性较弱,不容易被洗脱。按照英国药典及美国药典原料测定色谱条件,结果辅料对杂质A测定有干扰。按照美国药典片剂测定条件,辅料对杂质测定无干扰,但由于美国药典未收载杂质D,因此在该色谱条件下杂质D很难被洗脱,保留时间约102 min。因此,参照美国药典片剂色谱条件进行优化,采用了先等度洗脱盐酸黄酮哌酯,再升高乙腈比例洗脱极性较弱的杂质,从而缩短了杂质保留时间并达到有效分离。USP 39原料和片剂分别采用240 nm及293 nm作为检测波长,本研究中采用双波长240 nm及293 nm同时检测供试品中各杂质量及杂质个数,表明各杂质在这2个波长下响应基本一致,但293 nm基线更平稳,因此选择293 nm作为本品有关物质检测波长。盐酸黄酮哌酯片6个生产企业处方采用的辅料种类较多,各不相同,所选流动相可避免辅料干扰,同时保证主峰与各杂质峰分离度达到要求。
3.2 破坏试验
国内主要原料供应商均采用以3-甲基黄酮-8-羧酸为起始原料的合成工艺,该合成路线的主要杂质为3-甲基黄酮-8-羧酸、3-甲基黄酮-8-羧酸甲酯、3-甲基黄酮-8-羧酸乙酯。3-甲基黄酮-8-羧酸既是起始原料,也是主要降解产物,同时也是体内降解主要产物。因此,其是盐酸黄酮哌酯的关键杂质,另外,3个杂质是由3-甲基黄酮-8-羧酸与甲醇或乙醇或异丙醇成酯产生,可能在原料精制工序与精制溶剂发生反应产生,也可能在制剂过程中与溶剂反应产生。盐酸黄酮哌酯性质较稳定,原料在光照、酸、氧化、高温、高湿等条件下均较稳定,不易降解产生杂质,但在碱性条件下易降解产生杂质A。因此,本品在生产和贮藏过程中尽量避免碱性物质的影响。
3.3 杂质谱及杂质限度确定
对检出的主要杂质用国外药典对照品和高效液相色谱-质谱(HPLC-MS)进行了结构确认,确认检出的主要杂质为3-甲基黄酮-8-羧酸(杂质A)、3-甲基黄酮-8-羧酸甲酯(杂质 B)和 3-甲基黄酮 -8-羧酸乙酯(杂质C),与依据原料合成过程中推论出的杂质基本一致。6个厂家107批样品中均未检出杂质3-甲基黄酮-8-羧酸乙酯(杂质D),破坏试验中也均未检出杂质D,因此建议质量标准中不控制杂质D。F厂有2批样品杂质C含量很高,推测可能是原料或制剂生产工艺出现异常引起,具体原因有待进一步研究。考虑到目前国内生产企业质量控制是以中国药典规定为准,因此综合各国药典规定和样品检验结果,暂将限度定为杂质A不得超过0.5%,杂质B不得超过0.1%,杂质C不得超过0.15%,其他单个杂质不得超过0.1%,各杂质总和不得超过1.0%。
[1]张志根,孙百鸣,黄以政,等.盐酸黄酮哌酯治疗膀胱刺激症状[J].中华泌尿外科杂志,1996,17(10):638.
[2]简百录,刘晓辉,邓小枫,等.盐酸黄酮哌酯治疗前列腺痛的临床观察[J].西南国防医学,2005,15(2):163-164.
[3]赵德建,董其勇.盐酸黄酮哌酯治疗膀胱刺激征的临床疗效观察[J].中国药业,2001,10(10):51-52.
[4]刘 涛,李 猛,陈 平,等.盐酸黄酮哌酯在输尿管支架术后双“J”管综合征中的应用[J].中国临床研究,2013,26(10):1061-1062.
[5]鞠富霞,王金善,张京东,等.盐酸黄酮哌酯丙咪嗪联合治疗儿童不稳定膀胱合并遗尿症的临床研究[J].中国行为医学科学,2005,14(5):430.
[6]轩若亮,莫党生.盐酸黄酮哌酯加阿托品治疗157例输尿管结石性肾绞痛疗效观察[J].药物与临床,2005,34(4):604.
[7]徐晓云.奥尔芬加盐酸黄酮哌酯在输尿管结石性肾绞痛中的治疗作用[J].海南医学院学报,2005,11(6):530-531.
[8]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2015:1070-1072.
[9]王 玉,王铁杰,张廷兰.反相离子对高效液相色谱法测定盐酸黄酮哌酯片的含量[J].药物分析杂志,2002,22(3):202-205.
[10]The United States Pharmacopieial Convention.USP39-NF34[M].Rockville:The United States Pharmacopieial Convention,2015:1693,3914-3916.
[11]British Pharmacopoeia Commission.BP 2015[M].London:The Stationery Office,2015:1692-1693.
猜你喜欢
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
建材发展导向(2021年7期)2021-07-16
建材发展导向(2021年24期)2021-02-12
艺术品鉴(2020年6期)2020-12-06
中成药(2019年12期)2020-01-04
建材发展导向(2019年5期)2019-09-09
世界农药(2019年2期)2019-07-13
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
