
不同刈割强度下羊草转录组研究
2018-03-04 18:18赵劲博侯向阳武自念任卫波胡宁宁郭丰辉马文静中国农业科学院草原研究所内蒙古呼和浩特010010农业部草地生态与修复治理重点实验室内蒙古呼和浩特010010
草业学报 2018年2期
赵劲博,侯向阳*,武自念,任卫波,胡宁宁,郭丰辉,马文静(1.中国农业科学院草原研究所,内蒙古 呼和浩特 010010;2. 农业部草地生态与修复治理重点实验室,内蒙古 呼和浩特 010010)
羊草(Leymuschinensis)属于禾本科赖草属多年生根茎型草本植物,是欧亚大陆温带草原东部的重要建群种之一[1],具有抗旱、抗寒,耐盐碱、耐贫瘠等特点,在维持中国北方草原生态系统稳定、生物多样性、人工草地改良等方面有着重要意义。羊草持绿期长、叶量多,适口性好,营养价值高,其粗蛋白含量明显高于其他禾本科牧草[2],为多种牲畜喜食的高质量牧草。此外,羊草草地还是很好的割草地,具有很强的再生性,可为牲畜越冬提供优良饲草。
刈割是草地利用的重要方式之一,也是人为影响草地生态环境的重要因素。刈割减少了植物光合作用的面积,降低了碳和营养物质的积累,直接影响草原生产力[3]。刈割制度对饲草的再生生理和形态都会产生强烈的影响[4],而不同刈割强度会对刈割效果和牧草再生状态产生不一样的作用,刈割留茬高度与牧草的干草产量与损耗率有着直接的关系[5]。有研究表明,随着刈割强度的增大,羊草生物量、高度和密度呈下降趋势[6]。比起一些较为矮小的草原植物,羊草对刈割具有更强的敏感性,程度较大的机械损伤对其生长可造成更严重的影响[7]。刈割高度对羊草群落的影响在生理生态上研究较为广泛,但在个体的分子水平上还涉及甚少。
自从人们发现RNA在基因组和蛋白组研究中的关键作用,转录本识别和基因表达量计算便逐渐成为了分子生物学中重要研究方法[8]。随着高通量测序技术的不断完善,转录组数据挖掘与分析方法日趋成熟,一些没有基因组数据支持而又有着很高经济与生态价值的重要物种可以在测序成本和时间相对较少的条件下得到大批差异基因的表达情况,极大地帮助了相关物种生物信息数据库的完善,为基因克隆验证与表型研究提供了充足原始资料。在羊草低温、干旱、盐碱、刈割、放牧等胁迫作用下的转录组研究中,一些抗性基因与关键通路的挖掘与分析,为改良草地培育优质牧草奠定了基础[9]。羊草刈割与创伤的相关基因表达谱分析结果表明,刈割激活了茉莉酸生物合成途径基因的表达,诱导创伤信号,但核糖体蛋白基因、细胞分裂和膨胀相关基因及木质素生物合成基因表达却受到了抑制,影响羊草早期的恢复生长,而创伤处理下羊草脯氨酸合成的基因上调,引起脯氨酸的积累[10]。在对羊草刈割伤口对牛血清蛋白(BSA)响应的转录组研究中,有关细胞氧化、细胞程序性死亡、叶片卷曲动力和刈割创伤修复的基因有特异表达[11]。不同刈割处理对羊草的生长和利用产生重要影响,不同刈割强度转录组研究对于了解羊草响应刈割的关键分子机制及关键基因挖掘与应用有指导性意义。
本研究通过羊草在留茬高度分别为0、2、4、8、12 cm这5个不同强度刈割后再生叶片的转录组测序,RNA-seq数据组装,注释,以及对差异基因查找、筛选和功能与代谢途径富集分析,挖掘刈割相关的基因,并且根据基因表达量与刈割强度的相关性,提取出表达量随刈割强度的增强而持续升高或降低的基因。通过分析与刈割有关的关键基因,从分子层面探索了羊草响应不同程度机械损伤的机理,丰富了植物抗逆基因的数据库,为刈割对羊草在生理生化方面的影响提供研究方向,而且为培养牧草及其他经济作物的耐刈优良品种奠定理论基础,从而为改善生态环境、发展人工草地、提高农牧业生产力提供帮助。
1 材料与方法
1.1 试验材料
试验材料于2014年5月采集于中国农业科学院草原研究所锡林浩特放牧试验站,(44°15′ N,116°42′ E,海拔1100 m),小心将单株羊草地上部分连同地下根部挖出,移至培养室25 ℃,光照16 h,黑暗8 h条件下土培(营养土∶蛭石=3∶1)扩繁,挑选生长大小一致、状况良好的分蘖株移入多个盆中,每盆6~10株,培养备用。
1.2 试验设计
2016年1月开始,对分蘖株沿土壤表面刈割,进行室内土培并控制培养条件,保证各单株处于同一发育时期,同一生长环境。当植株生长14 d后(分蘖期),挑选生长状况一致的30盆植株使用直尺与剪刀进行梯度刈割处理并取样:刈割植株地上部分,分别留茬0,2,4,8,12 cm,依次记为A,B,C,D,E五组,以及不刈割对照组O,刈割3 d后取刈割组地上再生叶片以及对照组的全部叶片,每组6~10株植株(单盆)混合取样保证测序上样量,取样后立即液氮速冻,-80 ℃保存备用。
1.3 RNA提取及转录组测序
采用TRIzol法提取羊草叶片总RNA,并用琼脂糖凝胶电泳、Nanodrop 2000分析RNA的纯度和完整性,样品检测合格后,送至北京诺禾致源生物信息科技有限公司采用Illumina HiSeq-PE150进行测序。
1.4 数据组装与蛋白编码框预测
获得原始数据后,去除带接头(adapter)的reads;去除N(N表示无法确定碱基信息)的比例大于0.1%的reads;去除低质量reads(质量值Qphred≤20的碱基数占整个reads的50%以上的reads)。将过滤后的clean data使用软件Trinity[12]进行无参组装,设置计算最小kmer的参数(--min_kmer_cov)为2,最短contig长度(--min_contig_length)为200 bp,组装得到转录本,挑出最长转录本作为Unigene,随后使用软件TransDecoder[13]预测组装结果的蛋白编码框并将得到的蛋白序列用于注释。
1.5 组装结果注释与功能通路富集
通过使用比对软件BLAST[14]将得到的羊草转录本核酸序列和蛋白序列与蛋白核酸数据库进行比对(E≤10-5),每条序列挑取最好的比对结果使用Trinotate软件合并,得到了Unigene的注释结果。用到的数据库有:Nr(non-redundant protein database,非冗余蛋白数据库)、SwissProt(SwissProt protein database,蛋白质序列数据库)、Pfam[15](protein families database,蛋白质家族域数据库)、KEGG[16](Kyoto Encyclopedia of Genes and Genomes,东京基因与基因组百科全书)、Uniref90[17](全球蛋白资源数据库参考资料库)、KOG(Clusters of Orthologous Groups from 7 eukaryotic complete genomes,7个真核生物蛋白质直系同源数据库)。通过在线软件WEGO[18]将得到的GO进行统计分类,使用GOseq[19]以所有Unigene的GO注释结果为背景对上下调差异基因做GO功能富集分析。利用本地软件KOBAS 2.0[20]以所有Unigene的KEGG注释结果为背景对所有显著差异基因的KEGG代谢通路做富集分析。
1.6 差异基因与梯度基因的筛选
首先将测序得到的各处理clean reads利用比对软件Bowtie[21]分别回贴到所有Unigene上,并通过RSEM[22]计算每条Unigene在测序reads上的表达量。不同处理两两之间的显著性差异基因利用DEGseq[23]软件包根据MA-plot方法与随机抽样模型MARS(Random Sampling model)[23]筛挑得到,设定的差异表达倍数的对数(log2fc)大于1,P值小于10-3。
梯度基因筛挑方法:先提取所有处理两两之间的显著差异基因,再将其标准化后的表达量矩阵输入到用于研究处理梯度样本表达量变化的聚类软件stem[24]中,设定最大聚类数(maximum number of model profiles)为50,不同梯度单元之间最大变化(maximum number of units a model profile)为2,从而得到与刈割高度有关的梯度差异基因。
2 结果与分析
2.1 羊草转录组数据组装
Illumina高通量测序平台(HiSeq-PE150)测序共得到139767803条原始reads,总长度为41.94 Gb,去除接头和低质量片段后reads数为137626643条,大小为41.30 Gb,原始测序数据已上传至NCBI(SRX2792325)。使用Trinity对所有6个样品的转录组clean reads进行无参组装,共得到转录本270207条,总长度为191632770 bp(191.6 Mb),平均长度为709.21 bp,N50为1075 bp(1 kb)。提取最长转录本得到Unigene共180770条,总长度为106133700 bp(106.1 Mb),平均长度为587.12 bp,N50为806 bp。转录本与Unigene长度分布如图1所示。

图1 转录本与Unigene长度分布Fig.1 Length distribution of transcripts and Unigenes横坐标为转录本或Unigene的长度,纵坐标为其数目。 Abscissa for the length of transcripts or Unigenes, ordinate for their number.
2.2 Unigene注释结果
通过对Unigene的蛋白预测,得到具有蛋白编码框的Unigene共54555条,然后使用Blastx与Blastp将核酸及蛋白序列与数据库进行比对,总共注释到Unigene蛋白48097条,其中注释到SwissPort的有32913条(60.33%),Nr有46765条(85.72%),KOG有38470条(70.52%),Pfam有27219条(49.89%),GO有28409条(52.07%),KEGG有46461条(85.16%),未注释出的蛋白有6458条(11.84%)。KOG分类结果主要与信号转导机制(signal transduction mechanisms)、翻译后修饰和蛋白反转、分子伴侣(posttranslational modification, protein turnover, chaperones)等相关(图2)。有关细胞、细胞组分、结合、催化、细胞过程、代谢过程等结果在GO分类中较为显著(图3)。

图2 KOG功能分类Fig.2 KOG function classification横坐标为KOG种类,纵坐标为基因数目所占百分比。 Abscissa for KOG classification, ordinate for percent of genes number.

图3 GO分类Fig.3 GO classification横坐标为GO种类,纵坐标为基因数目所占百分比。 Abscissa for GO classification, ordinate for percent of genes number.
2.3 差异基因数目分析
根据羊草刈割不同高度的表达量矩阵,找到每个刈割高度处理的实验组(A、B、C、D、E)与不刈割对照组(O)表达量共有显著差异的基因24829个,其中A(留茬0 cm)有11618个,上调基因有7867个,下调基因有3751个;B(留茬2 cm)有9479个,上调基因6190个,下调基因3289个;C(留茬4 cm)有9159个,上调基因6031个,下调基因3128个;D(留茬8 cm)有13375个,上调基因10415个,下调基因2960个;E(留茬12 cm)有7143个,上调基因2777个,下调基因4366个。5个实验组与对照组共有的显著差异基因2579个,其中上调表达994个,下调表达1585个,这些基因极有可能与羊草响应刈割有关。
通过使用DEGseq对不同留茬高度处理样本进行两两比较分析,共找到30231个显著差异表达基因。然后利用stem软件对这些基因进行聚类,得到随着刈割强度增强(留茬高度降低),表达量随之规律变化的差异基因。其中,表达量呈上升趋势模式的差异基因有3499个(P=0)(图4中39号,图5左);表达量呈下降趋势模式的差异基因有1245个(P=0)(图4中8号,图5右);表达量呈先上升后下降趋势模式的差异基因有338个(P=5×10-8)(图4中48号);表达量呈先下降后上升的模式结果不显著(图4中9号)。

图4 所有差异基因stem聚类Fig.4 Stem clustering of all DEGs 曲线代表差异基因随胁迫程度增加表达量变化趋势,彩色框与无色框分布代表显著性和非显著性聚类,左上角与左下角数字分别为趋势类型编号和聚类显著性。 Curve for DEGs expression changed trend with the increase of stress degree. Color and colorless box represent significant and nonsignificant clustering respectively. The number of top left and lower left corner represent trend type and clustering significant respectively.

图5 随刈割强度增强表达量呈上升与下降趋势差异基因热图Fig.5 Heatmap of DEGs having up and down trend expression with the increase of mowing intensity左图呈上升趋势,右图呈下降趋势,表达量均经过z-score标准化处理。 Left represents up trend expression, right represents down trend expression, expression are normalized by z-score.
2.4 羊草刈割差异基因功能与代谢通路富集分析
将所有GO注释结果作为参考背景,将刈割实验组与不刈割对照组上调和下调显著性差异基因的GO分别进行富集处理,设定FDR小于0.05,得到刈割上调基因GO富集结果19条(表1)。在细胞组分方面,上调基因与胞外区、质外体、叶绿体有关;在生物途径上与碳水化合物代谢、损伤应答、过氧化氢分解有关;在分子功能上与水解酶活性、脂肪酸结合、蛋白二硫氧化还原酶活性有关。刈割下调基因GO富集结果24条(表2),在细胞组分上主要与膜有关;在生物途径上与跨膜运输、蛋白磷酸化、水杨酸反应有关;在分子功能上与蛋白激酶活性、多糖结合、跨膜转运体活性有关。

表1 刈割上调差异基因GO富集结果(FDR<0.05)Table 1 GO enrichment of mowing up-regulated DEGs (FDR<0.05)
FDR:错误发现率 False discovery rate.
利用KOBAS对差异基因KEGG代谢通路进行富集,得到BH法(Benjamini and Hochberg)校正P-value小于0.05的4条显著代谢通路(表3),分别为淀粉和蔗糖代谢、半乳糖代谢、苯丙烷生物合成、植物激素信号转导。其中,在植物激素信号转导方面,主要涉及脱落酸代谢中的脱落酸受体PYR/PYL家族(abscisic acid receptor PYR/PYL family)、蛋白磷酸酶2C(protein phosphatase 2C);茉莉酸代谢中茉莉酮酸酯ZIM结构域蛋白(jasmonate ZIM domain-containing protein)、茉莉素信号受体蛋白COI1(coronatine-insensitive protein 1);水杨酸代谢中病程相关蛋白1(pathogenesis-related protein 1)等。
此外,对刈割强度相关的梯度基因进行GO与KEGG富集,挑取GO富集结果(FDR<0.05)以及代谢通路富集结果(校正P-value小于0.05),发现表达量随刈割强度持续上升的基因主要与碳水化合物代谢、氨酰-tRNA合成、卟啉与叶绿素代谢、过氧化物酶体等有关(表4);而表达量随刈割强度持续下降的基因与蛋白激酶活性、叶绿体类囊体膜、光合作用、半胱氨酸与蛋氨酸代谢、硫代谢等有关(表5);表达量随刈割强度先上升后下降的基因与光合作用的天线蛋白(antenna proteins)、光捕获、光系统、膜组成、叶绿素等有关。

表2 刈割下调差异基因GO富集结果(FDR<0.05)Table 2 GO enrichment of mowing down-regulated DEGs (FDR<0.05)
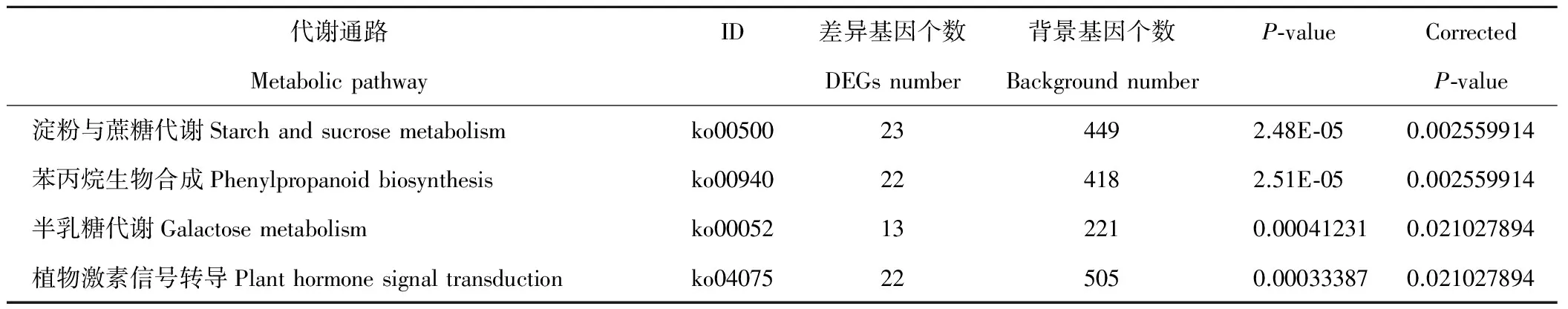
表3 刈割差异基因KEGG富集(校正P-value<0.05)Table 3 KEGG enrichment of mowing DEGs (Corrected P-value<0.05)
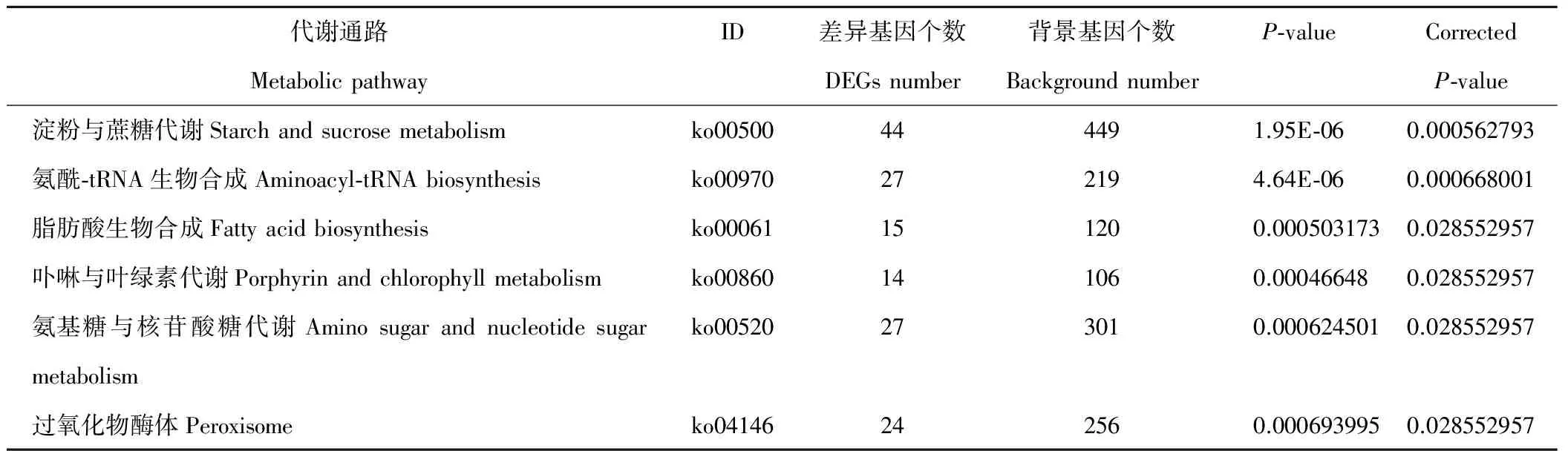
表4 表达量随刈割强度增强呈上升趋势差异基因KEGG富集(校正P-value<0.05)Table 4 KEGG enrichment of DEGs having up trend expression with the increase of mowing intensity (Corrected P-value<0.05)
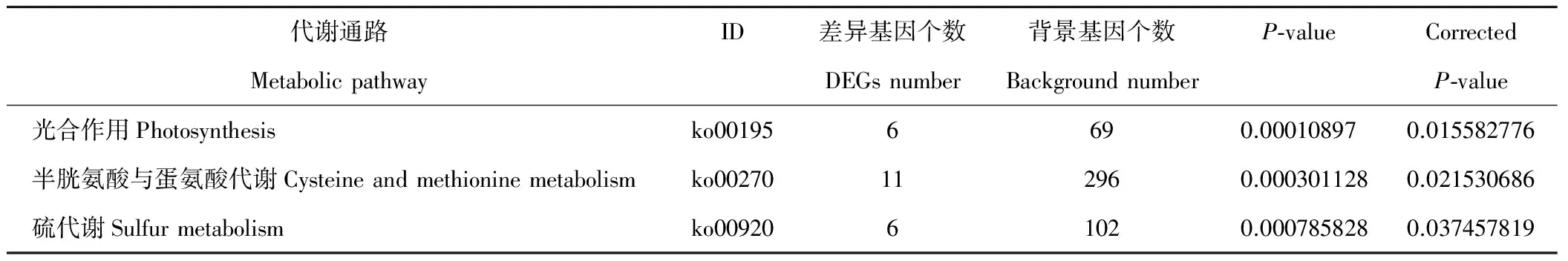
表5 表达量随刈割强度增强呈下降趋势差异基因KEGG富集(校正P-value<0.05)Table 5 KEGG enrichment of DEGs having down trend expression with the increase of mowing intensity (Corrected P-value<0.05)
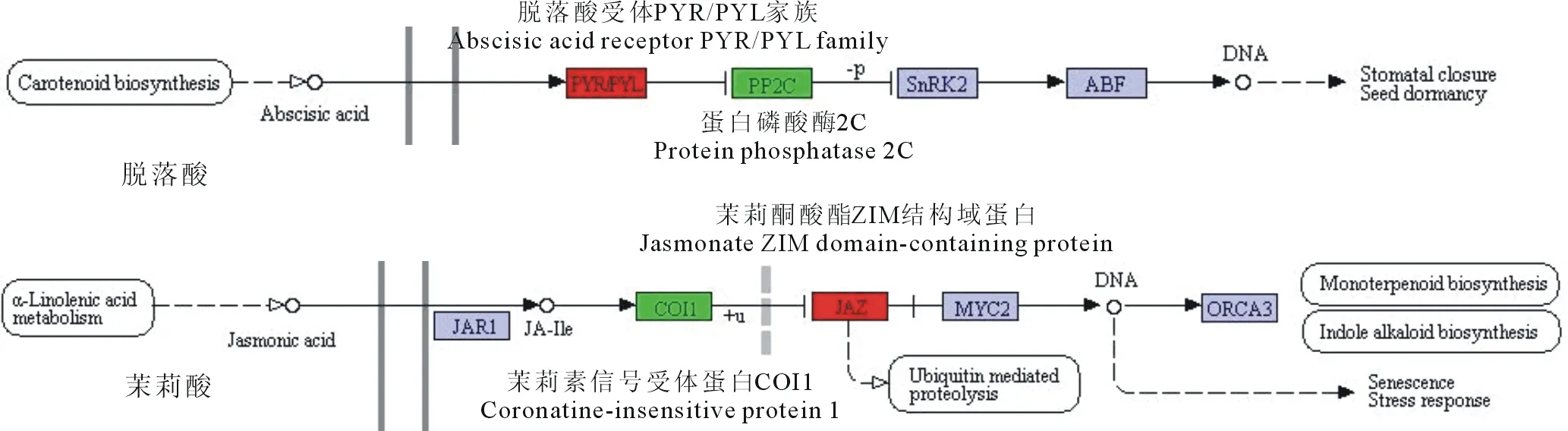
图6 刈割差异基因脱落酸与茉莉酸信号转导代谢通路富集结果Fig.6 ABA and JA signal transduction metabolic pathway enrichment of mowing DEGs红色代表上调,绿色代表下调。Red represents up-regulated, green represents down-regulated.
3 讨论
随着高通量测序技术的成熟与完善,越来越多无参考基因组的物种实现了转录组测序,并成功完成了在各种试验处理下物种转录表达水平变化的研究。羊草作为基因组数据庞大的异源四倍体草本植物,并且是一种具有抗寒抗旱耐盐耐刈等多种抗逆性的优良牧草,大量挖掘其关键功能基因是一项重要而艰巨的任务。目前与羊草抗逆相关的转录组研究已经取得了很多的突破性进展[10,25-26]。本研究通过不同刈割强度下羊草RNA转录表达情况,找到了与刈割相关的基因,并对其中部分基因的功能与代谢进行了进一步的分析研究。
刈割对植物在表型上造成可见的机械损伤,在其内部的分子水平上也会有不同程度的影响。在面对环境压力或生物胁迫时,植物会通过产生一些信号分子,在细胞内部发生关键的转录变化进而改变其生理状态[27],以应对外来的刺激与干扰。植物激素如生长素、脱落酸、茉莉酸、水杨酸等在植物抵御生物或非生物胁迫中起着重要作用。参与茉莉素形成的各种复合物广泛分布于植物中,影响着诸如花粉、果实形成,根系生长,茎叶卷曲等生长发育过程[28],茉莉素介导的抗性信号途径参与了有关机械损伤、病原菌侵染、昆虫噬咬等应对胁迫的抗逆反应[29]。COI1是一种茉莉素信号受体蛋白[30],可与其他多种蛋白组合形成SCFCOI1泛素连接酶复合体[31],从而促进茉莉素信号响应;JAZ蛋白家族是茉莉素信号途径中的一类抑制蛋白,是SCFCOI1泛素连接酶复合体的一类直接底物[32]。而本研究结果显示(图6)刈割处理导致COI1基因表达量下调,编码JAZ蛋白家族基因表达量上调,预测刈割对处理3 d后的羊草茎叶的茉莉素信号传递可能有抑制作用,从而对植物生长发育造成影响。Chen等[10]在羊草转录组试验的结果显示刈割处理后24 h内茉莉酸合成途径相关基因表达被激活,由此推测茉莉酸调控羊草在一定时间刈割胁迫下的抗逆反应可能主要依靠促进茉莉酸合成来实现,而茉莉素信号转导途径却不一定会被激活甚至是被抑制。脱落酸在植物种子萌发、芽的休眠、气孔开闭等生理过程中起着调控的作用,也介导了许多植物的环境逆境反应[33]。PYR/PYL/RCAR是ABA的一类受体蛋白[34-35],在脱落酸信号转导途径中起到正向调节的作用[36],其与脱落酸结合后,会使有负调控作用的PP2C蛋白活性被抑制[37],进而促进脱落酸信号转导。经过对数据的分析显示(图6),与不刈割相比,刈割处理使编码羊草PYR/PYL/RCAR蛋白家族的基因表达量呈现上调趋势,而编码PP2C蛋白的基因表达量下调,说明刈割可能会导致脱落酸更易与受体结合,进而促使脱落酸反应。从刈割上调差异基因的GO富集结果中得到了一些与损伤响应(GO:0009611)有关的基因,这些基因大多与拟南芥(Arabidopsisthaliana)应对损伤的基因有相似性,如参与乙烯生物合成的ACS6(At4g11280)基因和与衰老有关的SEN1(At4g35770)基因,前者在拟南芥机械损伤试验[38]下鉴定为非依赖于COI基因上调表达,后者为依赖COI基因而抑制表达,这与本试验结果COI1基因表达量下调相符,说明这两个基因与COI1基因的表达关系在羊草与拟南芥中可能是相同的。羊草刈割处理下的上调基因还比对到了拟南芥的FAR1(At5g22500)基因、CYP94C1(At2g27690)基因、TIFY11B(At1g72450)基因、WRKY40(At1g80840)基因、BSMT1(At3g11480)基因,这些基因在损伤处理下的拟南芥中也为上调表达[39-43]。
由于刈割强度的差别,可能导致植物体内相关基因的转录表达出现差异。随着刈割强度的增强,部分基因表达量呈现上升趋势,如超氧化物歧化酶(SOD)、过氧化氢酶(CAT)、过氧化物酶(POD)等与抗氧化酶活性有关的基因,这说明羊草在遭受刈割这种逆境胁迫后,会通过加强自身表达合成过氧化物酶的过程来清除有害物质,且刈割强度越高这种抗性越强,类似的结果在不同损伤高度处理冷蒿(Artemisiafrigida)的研究[38]中也有体现,该研究表示随着机械损伤强度增加,冷蒿叶片的SOD、CAT和POD活性也随之增加,可见植物会通过加强自身的抗氧化能力来适应更高强度的损伤胁迫。而部分基因则随着刈割强度增强表达量逐渐下降,如与光合作用有关的基因,这可能是由于刈割导致了羊草叶片减少,且留茬高度越低,其叶面积就越小,进而造成光合作用逐渐下降,相关基因的表达也受到了进一步限制,如与叶绿体类囊体膜、光系统Ⅱ复合体、光合电子传递链等结构与功能有关的基因表达量呈下降趋势。
本研究针对羊草刈割强度设置了不同留茬高度作为处理实验组,通过转录组测序得到大量的转录本数据。在常规分析的基础下,重点对注释到其他物种已知序列的关键基因进行了研究,而对于一些功能不明确或没有注释结果但表达量有规律性的基因,还有待在结构与功能等方面进行进一步的分析探索,从而使数据挖掘与利用更深入全面,以助于探求羊草在机械损伤逆境下最本质的机理。
4 结论
通过转录组测序与生物信息学分析,获得总长度达192.6 Mb,N50为1075 bp的转录本270207条,经过蛋白编码框预测与序列注释,共得到有注释结果的Unigene 48097条。得到刈割实验组与不刈割对照组上调基因994个,主要富集到了碳水化合物代谢过程、损伤应答、过氧化氢分解过程等功能;下调基因1585个,主要富集到了跨膜运输、蛋白磷酸化、水杨酸反应等功能。随刈割强度增强,表达量呈上升趋势的基因有3499个,富集分析结果显示刈割强度可能与氨酰-tRNA生物合成、脂肪酸生物合成、卟啉与叶绿素代谢、氨基糖和核苷酸糖代谢、过氧物酶体有关;随刈割强度增强,表达量呈下降趋势的基因有1245个,主要与光合作用、半胱氨酸和蛋氨酸代谢、硫代谢有关。
References:
[1] Li Y H. The divergence and convergence ofAneurolepidiumchinesesteppe andStipgrandissteppe under the grazing influence in Xilin river valley, Inner Mongolia. Chinese Journal of Plant Ecology, 1988, 12(3): 189-196.
李永宏. 内蒙古锡林河流域羊草草原和大针茅草原在放牧影响下的分异和趋同. 植物生态学报, 1988, 12(3): 189-196.
[2] Zhao H. The Study of Nutrition Value of TheLeymuschinensisfor Dairy Cattle and Assiociative Effects of Feeds. Daqing: Heilongjiang Bayi Agricultural University, 2008.
赵鹤. 羊草对奶牛营养价值及其日粮组合效应的研究. 大庆: 黑龙江八一农垦大学, 2008.
[3] Ferraro D O, Oesterheld M. Effect of defoliation on grass growth. A quantitative review. Oikos, 2002, 98(1): 125-133.
[4] Davidson J L, Milthorpe F L. Leaf growth in dactylis glomerate following defoliation. Annals of Botany, 1966, 30(2): 173-184.
[5] Wang Z F, Wang D J, Yu H Z,etal. Effects of cutting time and stubble height on hay yield and quality ofLeymuschinensismeadow. Pratacultural Science, 2016, (2): 276-282.
王志锋, 王多伽, 于洪柱, 等. 刈割时间与留茬高度对羊草草甸草产量和品质的影响. 草业科学, 2016, (2): 276-282.
[6] Zhong Y K, Bao Q H. The effects of different mowing intensity on natural grassland. Grassland of China, 1999, (5): 15-18.
仲延凯, 包青海. 不同刈割强度对天然割草地的影响. 中国草地, 1999, (5): 15-18.
[7] Han L, Guo Y J, Han J G,etal. A study on the diversity and aboveground biomass in aLeymuschinensismeadow steppe community under different cutting intensities. Acta Prataculturae Sinica, 2010, 19(3): 70-75.
韩龙, 郭彦军, 韩建国, 等. 不同刈割强度下羊草草甸草原生物量与植物群落多样性研究. 草业学报, 2010, 19(3): 70-75.
[8] Conesa A, Madrigal P, Tarazona S,etal. A survey of best practices for RNA-seq data analysis. Genome Biology, 2016, 17(1): 181.
[9] Hou X Y. Advances and prospects of grassland plant basic biology research. China Basic Science, 2016, 18(2): 67-76.
侯向阳. 草原植物基础生物学研究进展与展望. 中国基础科学, 2016, 18(2): 67-76.
[10] Chen S, Cai Y, Zhang L,etal. Transcriptome analysis reveals common and distinct mechanisms for sheepgrass (Leymuschinensis) responses to defoliation compared to mechanical wounding. Plos One, 2014, 9(2): e89495.
[11] Huang X, Peng X, Zhang L,etal. Bovine serum albumin in saliva mediates grazing response inLeymuschinensisrevealed by RNA sequencing. BMC Genomics, 2014, 15(1): 1126.
[12] Grabherr M G, Haas B J, Yassour M,etal. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 2011, 29(7): 644-652.
[13] Haas B J, Papanicolaou A, Yassour M,etal. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols, 2013, 8(8): 1494-1512.
[14] Altschul S F, Gish W, Miller W,etal. Basic local alignment search tool. Journal of Molecular Biology, 1990, 215(3): 403-410.
[15] Punta M, Coggill P C, Eberhardt R Y,etal. The Pfam protein families database. Nucleic Acids Research, 2012, 36: 263-266.
[16] Kanehisa M, Goto S, Sato Y,etal. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research, 2012, 40: D109-D114.
[17] Wu C H, Apweiler R, Bairoch A,etal. The universal protein resource (UniProt): an expanding universe of protein information. Nucleic Acids Research, 2006, 34: D187-D191.
[18] Ye J. WEGO: a web tool for plotting GO annotations. Nucleic Acids Research, 2006, 34(Web Server issue): W293-W297.
[19] Young M D, Wakefield M J, Smyth G K,etal. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology, 2010, 11(2): R14.
[20] Xie C, Mao X, Huang J,etal. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research, 2011, 39(Web Server issue): W316-W322.
[21] Langmead B, Trapnell C, Pop M,etal. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology, 2009, 10(3): R25.
[22] Li B, Dewey C N, Li B,etal. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 2011, 12(1): 323.
[23] Wang L, Feng Z, Wang X,etal. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics (Oxford, England), 2010, 26(1): 136-138.
[24] Ernst J, Barjoseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics, 2006, 7(7): 1-11.
[25] Sun Y, Wang F, Wang N,etal. Transcriptome exploration inLeymuschinensisunder saline-alkaline treatment using 454 pyrosequencing. Plos One, 2013, 8(1): e53632.
[26] Zhao P, Liu P, Yuan G,etal. New insights on drought stress response by global investigation of gene expression changes in sheepgrass (Leymuschinensis). Frontiers in Plant Science, 2016, 7(147): 954.
[27] Devoto A, Ellis C. Expression profiling revealsCOI1 to be a key regulator of genes involved in wound- and methyl jasmonate-induced secondary metabolism, defence, and hormone interactions. Plant Molecular Biology, 2005, 58(4): 497-513.
[28] Turner J G, Ellis C, Devoto A. The jasmonate signal pathway. Plant Cell, 2002, 14(Suppl 1): S153-S164.
[29] Zhou W. Molecular Mechanism of Jasmonate-regulated Plant Defense Controled by JAV1. Beijing: Tsinghua University, 2013.
周武. JAV1调控茉莉素介导的植物抗性反应的分子机理研究. 北京: 清华大学, 2013.
[30] Yan J, Zhang C, Gu M,etal. TheArabidopsisCORONATINE INSENSITIVE1 protein is a jasmonate receptor. Plant Cell, 2009, 21(8): 2220.
[31] Adams E, Turner J. Illuminating COI1: a component of theArabidopsisjasomonate receptor complex also interacts with ethylene signaling. Plant Signaling & Behavior, 2010, 5(12): 1682-1684.
[32] Thines B, Katsir L, Melotto M,etal. JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature, 2007, 448(7154): 661-665.
[33] Chen X Y, Tang Z C. Plant Physiology and Moleculer Biology. Beijing: Higher Education Press, 2012: 553-554.
陈晓亚, 汤章城. 植物生理与分子生物学. 北京: 高等教育出版社, 2012: 553-554.
[34] Ma Y, Szostkiewicz I, Korte A,etal. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science, 2009, 324(5930): 1064.
[35] Park S Y, Fung P, Nishimura N,etal. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science, 2009, 324(5930): 1068-1071.
[36] Gonzalez-Guzman M, Pizzio G A, Antoni R,etal.ArabidopsisPYR/PYL/RCAR receptors play a major role in quantitative regulation of stomatal aperture and transcriptional response to abscisic acid. The Plant Cell, 2012, 24(6): 2483-2496.
[37] Schweighofer, Alois, Hirt,etal. Plant PP2C phosphatases: emerging functions in stress signaling. Trends in Plant Science, 2004, 9(5): 236.
[38] Jia L, Liu M M, Zhang H Q,etal. Antioxidant defense system responses ofArtemisiafrigidato mechanical damage. Journal of Zhejiang A & F University, 2016, 33(3): 462-470.
贾丽, 刘盟盟, 张洪芹, 等. 冷蒿抗氧化防御系统对机械损伤的响应. 浙江农林大学学报, 2016, 33(3): 462-470.
[39] Domergue F, Vishwanath S J, Joubès J,etal. Three Arabidopsis fatty acyl-coenzyme A reductases, FAR1, FAR4, and FAR5, generate primary fatty alcohols associated with suberin deposition. Plant Physiology, 2010, 153(4): 1539-1554.
[40] Koo A J K, Cooke T F, Howe G A. Cytochrome P450 CYP94B3 mediates catabolism and inactivation of the plant hormone jasmonoyl-L-isoleucine. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(22): 9298.
[41] Yan Y, Stolz S, Chételat A,etal. A downstream mediator in the growth repression limb of the jasmonate pathway. Plant Cell, 2007, 19(8): 2470-2483.
[42] Walley J W, Coughlan S, Hudson M E,etal. Mechanical stress induces biotic and abiotic stress responses via a novel cis-element. Plos Genetics, 2007, 3(10): 1800-1812.
[43] Chen F, D’Auria J C, Tholl D,etal. AnArabidopsisthalianagene for methylsalicylate biosynthesis, identified by a biochemical genomics approach, has a role in defense. Plant Journal, 2003, 36(5): 577-588.
猜你喜欢
现代畜牧科技(2021年4期)2021-07-21
种子(2020年4期)2020-12-22
文苑(2020年12期)2020-04-13
心电与循环(2020年1期)2020-02-27
意林·全彩Color(2019年11期)2019-12-30
意林彩版(2019年11期)2019-11-22
江苏农业科学(2017年5期)2017-04-15
戏剧之家(2016年1期)2016-02-25
现代畜牧科技(2016年9期)2016-02-21
草业科学(2015年9期)2015-12-11
