
超高速液相色谱用纳米ZrO2晶体颗粒的制备及机理
2018-03-02 01:36海弘毅,苏宏久,王树东,刘彦军
大连工业大学学报 2018年1期
海 弘 毅, 苏 宏 久, 王 树 东, 刘 彦 军
( 1.大连工业大学 轻工与化学工程学院, 辽宁 大连 116034;2.中国科学院 大连化学物理研究所, 辽宁 大连 116023 )
0 引 言
ZrO2纳米材料是一种良好的耐腐蚀、耐磨损和耐高温的金属氧化物材料[1]。具有粒径分布窄、超细、非聚集、均匀、可控等优点的特定形貌的ZrO2化学组成材料已被应用于燃料电池电解质、氧传感器、介电材料、无机涂层、催化剂和色谱填充料等[2]材料制备当中,尤其是应用于超高速液相色谱柱材料的研究日趋受到关注。超高速液相色谱柱材料要求是亚微米到微米级的球形颗粒材料,具有较窄的尺寸分布。相比于标准、多孔的色谱柱材料,无孔液相色谱柱材料缺少粒子内的孔分布,其界面质量传递更快,能够有效地提高色谱分析时间。Stegeman等[3]已将亚微米无孔的颗粒用于色谱中含水大分子的分离。
目前,无孔、球形ZrO2纳米颗粒的制备方法主要有沉淀法、溶胶-凝胶法、溶剂热(水热)法和微乳液法[4-6]等。沉淀法、溶胶凝胶法、溶剂热(水热)法得到的颗粒尺寸分布范围广、易产生团聚,无法制备出均一、单分散的ZrO2颗粒。微乳液法由水和油两相形成的界面使溶液中能够形成油包水或者水包油的两相体系,进而形成一个特殊的球形微反应容器[7],在该反应容器中发生生成ZrO2的水解反应,可得到球形度高、分散性好、均一性好、无孔的纳米ZrO2颗粒。Tai等[8]制备出了球形ZrO2颗粒,但是球形度和分散性均较差。本研究采用微乳液法,可控制备球形度高、分散性优、均一性好、无孔的纳米ZrO2颗粒,对颗粒进行形貌和结构表征,并且初步对微乳液法的合成机理进行探讨。
1 实 验
1.1 试 剂
正丙醇锆,分析纯,Aldrich;正丁醇、硬脂酸,分析纯,国药集团;去离子水,自制。
1.2 单分散球形ZrO2颗粒的制备
向100 mL无水正丁醇中加入1 g硬脂酸,搅拌10 min,当硬脂酸完全溶解在无水正丁醇溶液中时,加入11.5 mL无水正丙醇锆溶液,同时搅拌10 min,形成透明溶液。将85 mL无水正丁醇溶液和1.5 mL去离子水混合,搅拌均匀,超声振荡1 min,将两种溶液混合,搅拌1 min,静止陈化150 min,分别用正丁醇和异丙醇清洗3次,抽滤得到初始样品。初始样品经过干燥、焙烧得到最终ZrO2样品。焙烧程序:室温升温至450 ℃,保持3 h,之后升温到750 ℃,保持5 h,升温速率为5 ℃/min,自然降温。
1.3 ZrO2颗粒表征
采用日本电子JSM-7800F场发射扫描电子显微镜表征样品的形貌,Malvern粒度仪表征样品颗粒粒径,荷兰帕纳科Xpert Pro XRD衍射仪表征样品的物相和晶型,NETZSCH Leading Thermal Analysis STA 449F3进行TG-DTA分析,美国康塔公司NOVA 2200e Surface Area & Pore Size Analyzer N2吸附仪测试样品的比表面积和孔体积。
2 结果与讨论
2.1 形貌和尺寸分析
图1为ZrO2粉末750 ℃焙烧之后的SEM图。可以看出,高温焙烧之后的ZrO2微球表面球形形貌保持完好,制备的ZrO2颗粒球形度高、分散性好,且ZrO2颗粒表面较为光滑。

图1 ZrO2粉体的SEM图
图2为ZrO2粉末的粒度分布曲线图,颗粒的平均粒径为1 080 nm,分散性指数PDI为0.191,均一性好。
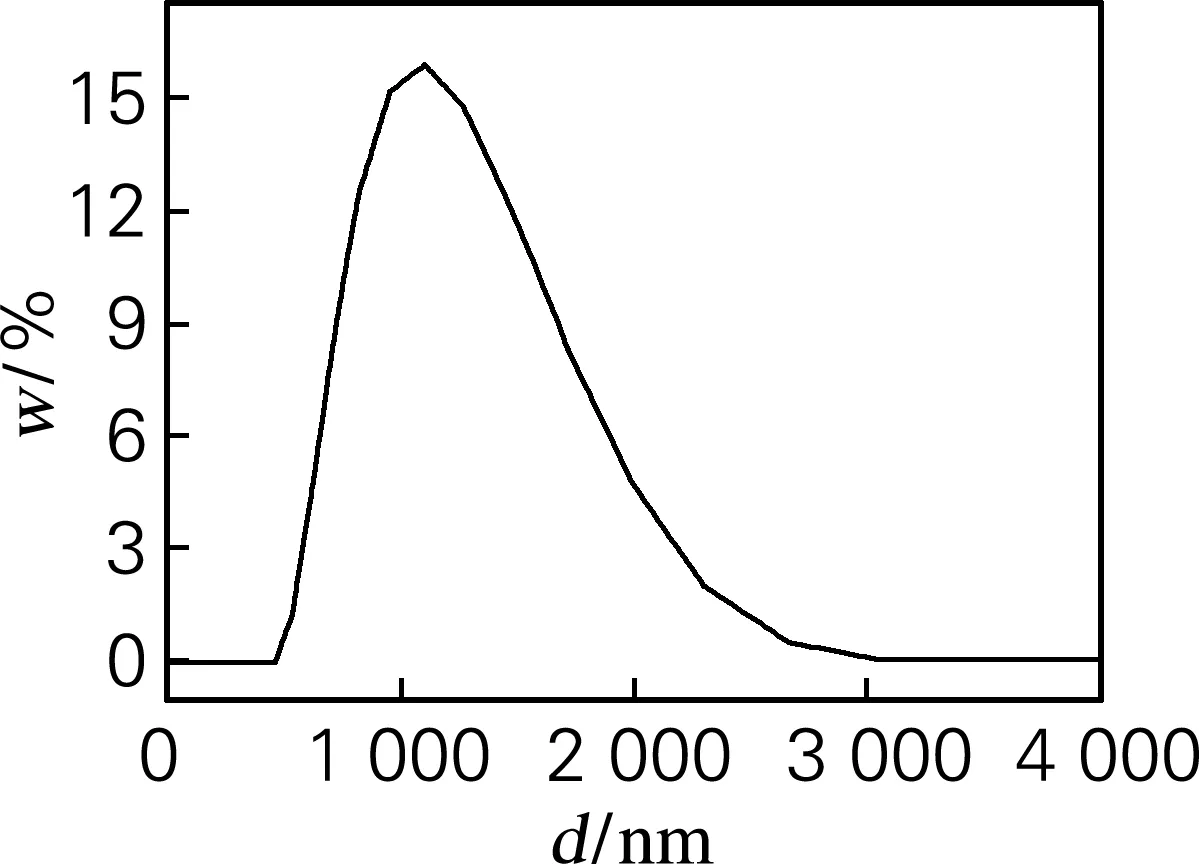
图2 ZrO2粉体的粒度分布曲线
球形ZrO2颗粒的形成通常认为经过两个步骤完成,如图3所示。首先在溶液中形成10 nm左右的ZrO2前驱体Zr(OH)4微小颗粒,微小颗粒团聚形成最终的球形颗粒。
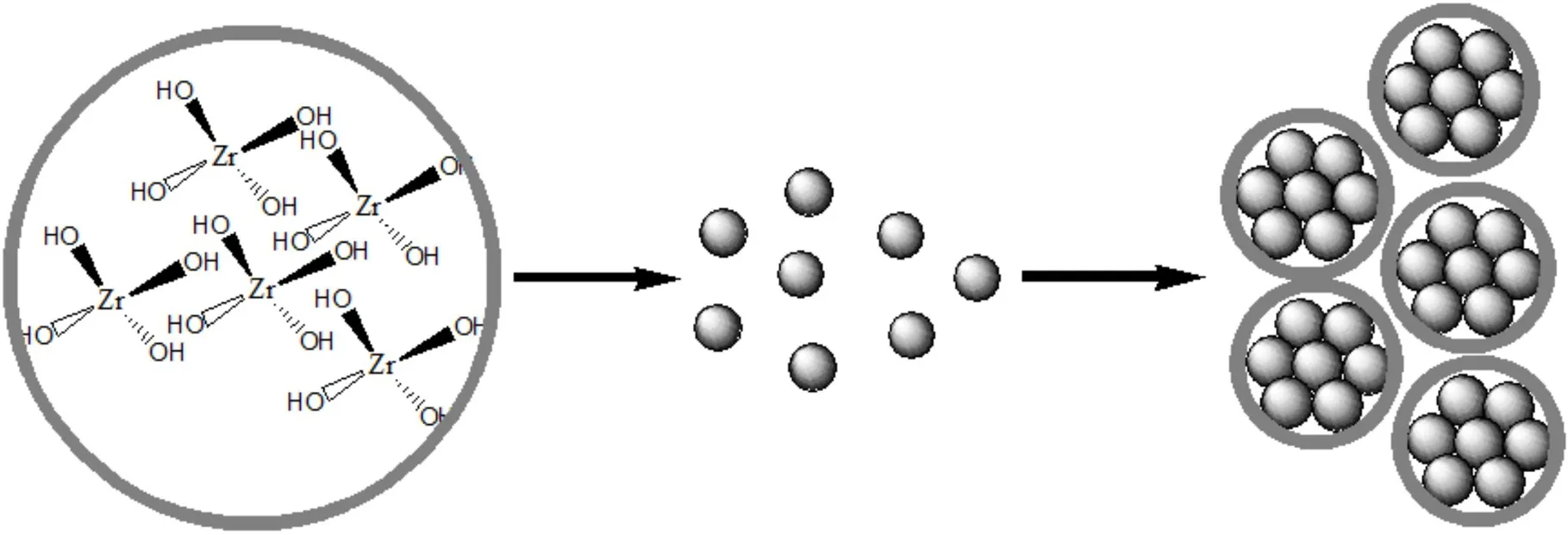
图3 锆醇盐水解形成的初始微小颗粒快速结合长大成球形示意图
Fig.3 Schematic process of the spherical particles formed from hydrolysis of the initial tiny zirconium alkoxide
Zr(OR)4+H2O→Zr(OH)4+4ROH
(1)
Zr(OH)4→ZrO2+2H2O
(2)
此外,含水的ZrO2之间相互聚合会产生Zr—O—Zr桥键,通过水解和浓缩形成大分子网状结构[9]。ZrO2纳米颗粒的形状和尺度由多种因素控制,例如合成方式、稳定剂、表面活性剂、反应物浓度、反应温度、搅拌速率和反应时间。本实验采用的微乳液法中,水相扩散到正丁醇的油相中,均匀分散后能够形成分散性较好的微小水相液滴,在微乳液体系当中,可以避免水相之间的相互接触,分散于油相中的水可以被认为是一种独立微小的反应器。
在曲面上的不同部位,曲面的弯曲方向及曲率各不相同,产生的附加压力的方向和大小也不同。在凸液面处附加压力指向液滴内部,而凹液面处附加压力的指向则相反,这种不平衡力迫使液滴自动调整形状,最终呈现球形,因为只有呈现球形,球面各点的曲率才相同,各处的附加压力也相同,液滴或气泡才会稳定存在[10]。在分散均匀的微小水介质球形反应器当中,ZrO2晶核形成之后快速生长,聚合形成球形ZrO2颗粒。
2.2 XRD物相分析
图4为颗粒在不同焙烧温度下的XRD图。可以看出,未经过焙烧的样品是无定型的,随着焙烧温度的升高,可以发现粉末颗粒的两种晶型转变。颗粒在500 ℃焙烧时,晶型由无定型转变为亚稳定四方形;在700 ℃焙烧时,晶型由亚稳定四方形向单斜型转变。
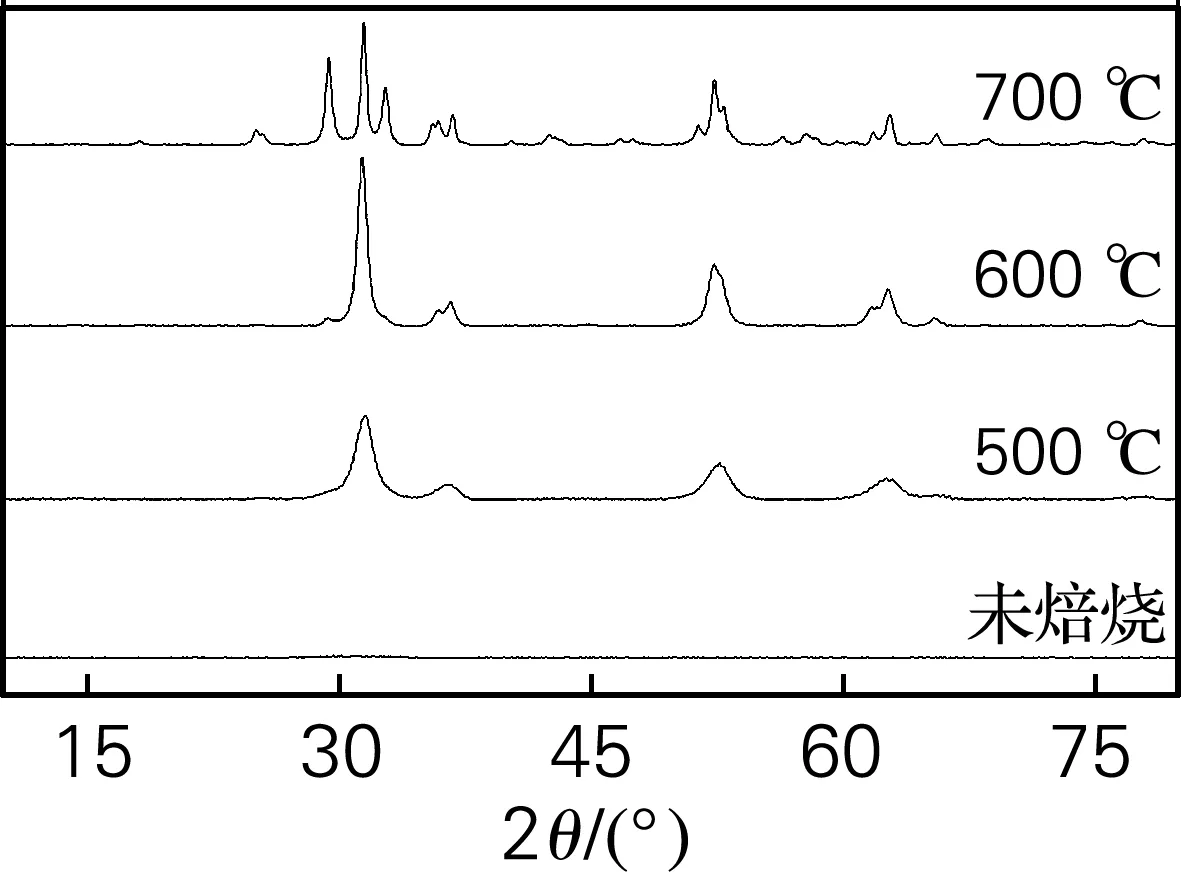
图4 ZrO2前驱体不同焙烧温度下的XRD图
造成上述转变的原因:由于较为少量的无定型相和亚稳定四方形相比较相似,初始晶体在500 ℃焙烧之后产生了亚稳定四方形,分析计算得到晶体颗粒平均尺寸为6.7 nm。一般可以认为10~20 nm为代表性的晶体临界尺寸,在临界晶体尺寸之下,由于聚合晶体表面能的不同,相对于单斜型,亚稳定四方形容易形成热动态平衡优先相,因此容易形成亚稳定四方形晶相。此外,在加热期间生成的网状连接结构可以阻碍ZrO2晶粒的生长,导致ZrO2晶粒处于纳米级别,由于纳米ZrO2颗粒的表面影响,大量的原子吸附在纳米晶体的表面上,产生大量氧空位,因此,在700 ℃ 以下会出现暂时性的亚稳定四方形晶体。随着温度升高,氧空位数量减少,造成亚稳定四方形ZrO2向单斜型ZrO2转变。因此,在高温焙烧之后,会形成稳定单斜型晶型的ZrO2颗粒。
Zr(OR)4+H2O→(OR)3Zr(OH)+R(OH)
(3)
(OR)3Zr(OH)+(OR)3Zr(OR)→
(OR)3Zr-O-Zr(OR)3+ROH
(4)
2.3 TG-DTA分析
图5为ZrO2前驱体颗粒加热至800 ℃的TG-DTA图。TG曲线中,前驱体颗粒在30~300 ℃ 有一个较宽的放热峰和大量的质量缺失,质量变化为16.18%,这主要是由于物理吸附的结合水分和一些溶剂蒸发所造成的。300~750 ℃ 颗粒的质量变化为26.37%,大约在350和450 ℃分别出现两个明显的放热峰,前者可能是由于附着在颗粒表面的有机物的氧化和分解造成的,后者是由于无定型ZrO2晶体向亚稳定四方形转变所造成的。在700 ℃左右有一个不明显的放热峰,可能是由于亚稳定四方形的ZrO2晶体向单斜型晶体转变所造成的。
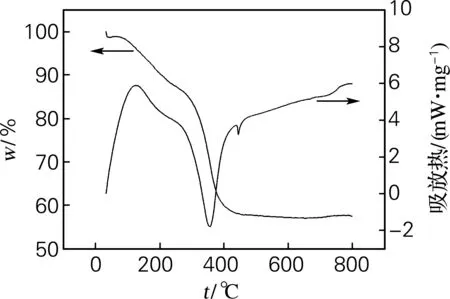
图5 ZrO2前驱体粉末的TG-DTA曲线
通过对ZrO2前驱体Zr(OH)4进行焙烧,使其脱去水分子和所吸附的有机物从而形成晶体,且晶体由无定型转变为亚稳定型,最后转变为稳定晶型的晶体颗粒。这一转变过程对ZrO2晶体的制备非常必要。
2.4 比表面积和孔分析
图6为ZrO2前驱体颗粒Zr(OH)4在60 ℃真空干燥之后的吸附-脱附曲线。采用N2吸附-脱附曲线的BET方法测得颗粒的比表面积为6.314 m2/g。从吸附量和孔分析计算可以看出,样品几乎没有孔体积,属于无孔的ZrO2球。
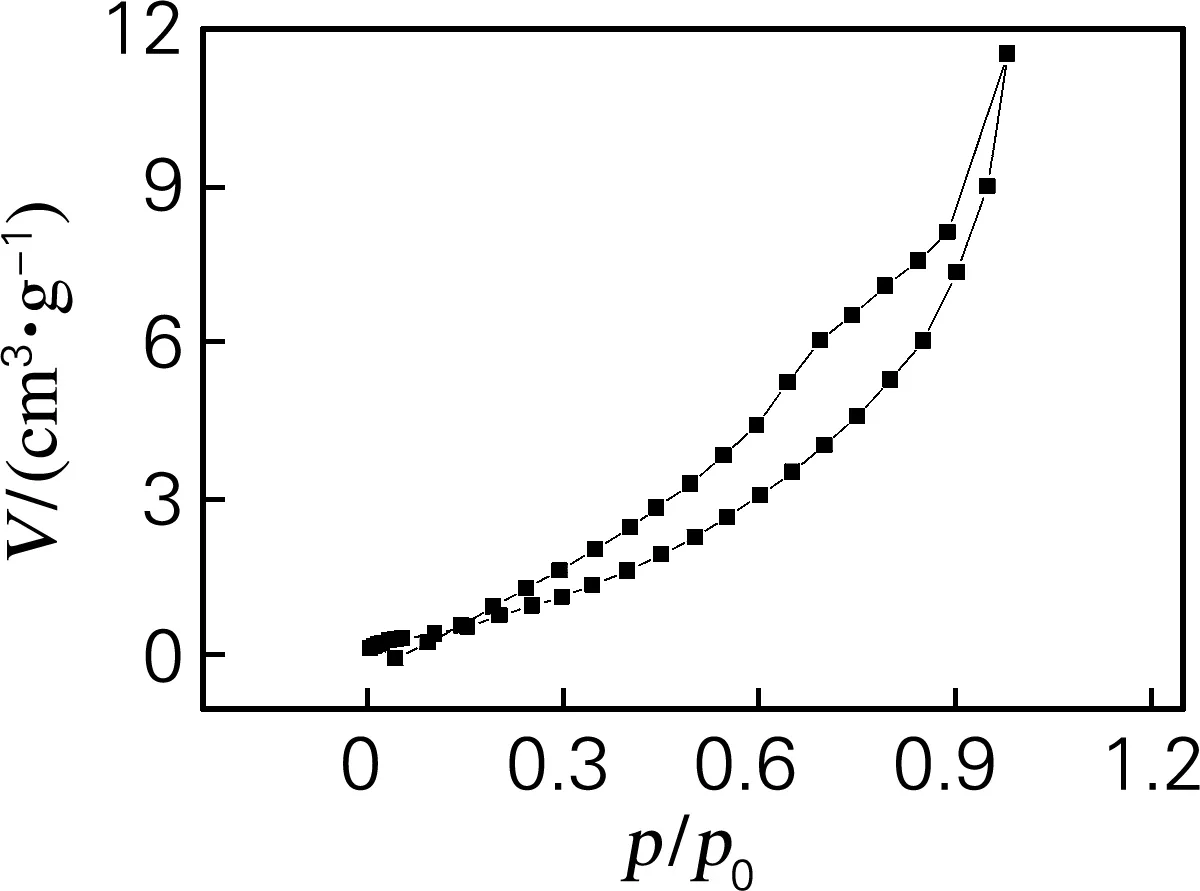
图6 ZrO2前驱体颗粒没有焙烧时的N2吸附-脱附曲线
Fig.6 N2adsorption-desorption curves of ZrO2precursor without calcination
图7是经过750 ℃焙烧之后ZrO2的N2吸附-脱附曲线。颗粒的比表面积为3.338 m2/g,且通过吸附量和孔分析可以表明样品也几乎没有孔结构。样品焙烧前后均呈现出无孔的结构,表明ZrO2颗粒表面硬脂酸的吸附不会导致孔的出现,颗粒致密堆叠形成无孔颗粒。但是,在焙烧之后,样品的吸附量和比表面积均减小,这主要是由于ZrO2晶体颗粒从四方形向单斜型转变时,会伴随有3%~5%的体积扩展,体积扩展会导致部分孔消失。制备的ZrO2纳米颗粒满足超高速液相色谱柱材料无孔结构的要求。
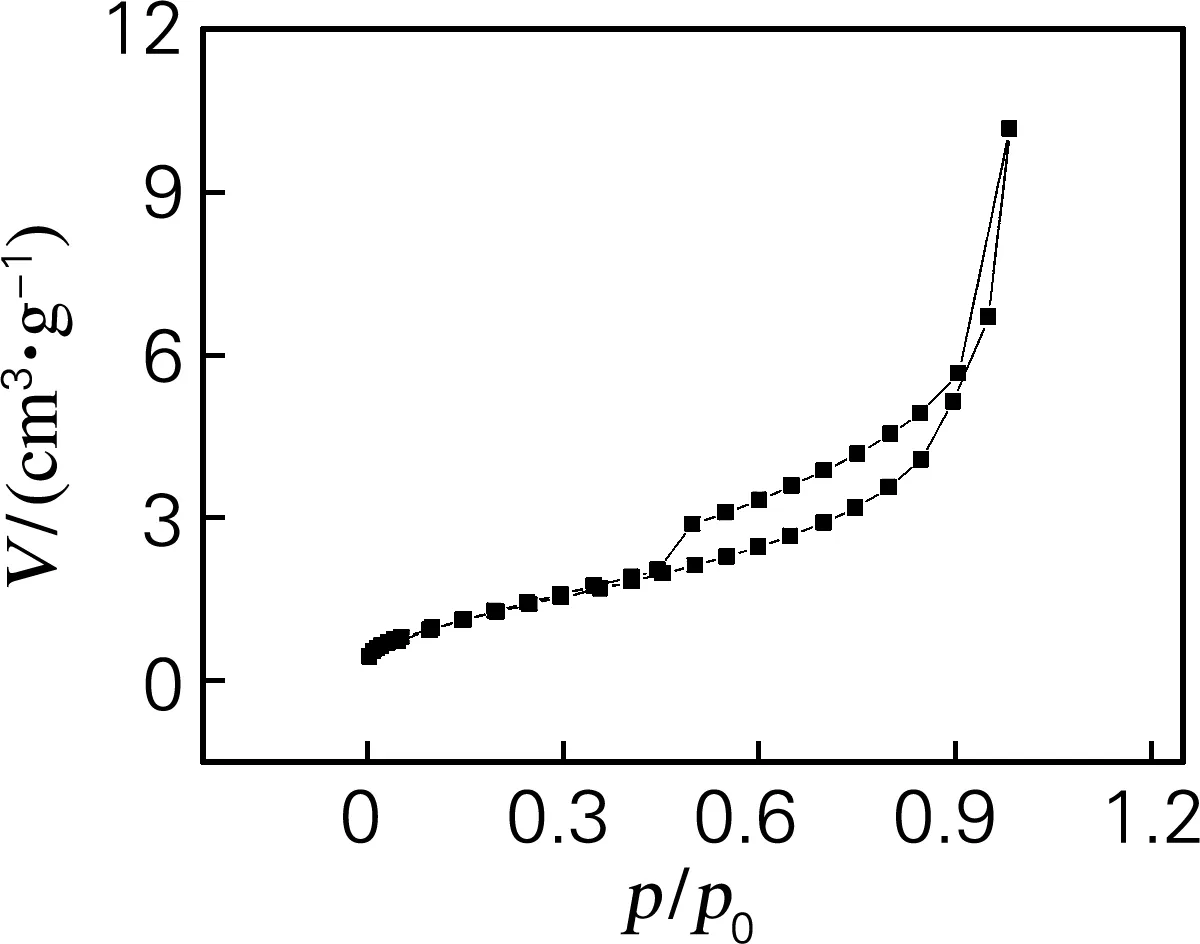
图7 ZrO2前驱体颗粒750 ℃焙烧后的N2吸附-脱附曲线
Fig.7 N2adsorption-desorption curves of ZrO2precursor calcination at 750 ℃
3 结 论
以水、正丁醇混合微乳液体系作为反应相,通过正丙醇锆、正丁醇混合溶液与反应相发生水解反应,制备得到了颗粒尺寸约1 000 nm的ZrO2粉末。与其他制备方法相比,采用具有水油相界面的微乳液法制备得到的ZrO2颗粒具有无团聚、粒径均一和球形度高的优点。经过高温焙烧之后,样品粉末由无定型转变成亚稳定四方形,之后由亚稳定四方形转变成为固定单斜型晶相。N2吸附-脱附表征发现,制备的球形ZrO2颗粒粉末的比表面积较低,几乎没有孔结构。制备的无孔、球形ZrO2纳米颗粒满足超高速液相色谱柱超快分离材料的要求。
[1] NGUYEN T-D. From formation mechanisms to synthetic methods toward shape-controlled oxide nanoparticles[J]. Nanoscale, 2013, 5(20): 9455-9482.
[2] ZHAO N N, PAN D C, NIE W, et al. Two-phase synthesis of shape-controlled colloidal zirconia nanocrystals and their characterization[J]. Journal of the American Chemical Society, 2006, 128(31): 10118-10124.
[3] STEGEMAN G, OOSTERVINK R, KRAAK J C, et al. Hydrodynamic chromatography of macromolecules on small spherical non-porous silica particles[J]. Journal of Chromatography A, 1990, 506(1): 547-561.
[4] GARG N, MITTAL V K, BERA S, et al. Preparation and characterization of tetragonal dominant nanocrystalline ZrO2obtained via direct precipitation[J]. Ceramics International, 2012, 38(3): 2507-2512.
[5] CHANG Y L, WANG C, LIANG T X, et al. Sol-gel synthesis of mesoporous spherical zirconia[J]. RSC Advances, 2015, 5(127): 104629-104634.
[6] 王晶,许吉泰,龚念.水热法制备球形二氧化锆粉体的研究[J].硅酸盐通报,2013(5):936-940.
[7] HUANG Y, MA T N, YANG J L, et al. Preparation of spherical ultrafine zirconia powder in microemulsion system and its dispersibility[J]. Ceramics International, 2004, 30(5): 675-681.
[8] TAI C Y, HSIAO B Y, CHIU H Y. Preparation of spherical hydrous-zirconia nanoparticles by low temperature hydrolysis in a reverse microemulsion[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2004, 237(1/2/3): 105-111.
[9] JOHANNA W, STEFANIE E A, GEORG M. Synthesis and characterisation of monodispersed zirconia particles[J]. European Journal of Inorganic Chemistry, 2005(15): 3149-3155.
[10] 傅玉普,郝策,蒋山.多媒体CAI·物理化学[M].大连:大连理工大学出版社,2004.
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
中成药(2018年9期)2018-10-09
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
材料科学与工程学报(2016年1期)2017-01-15
优雅(2016年2期)2016-06-03
当代化工研究(2016年7期)2016-03-20
中国果菜(2016年9期)2016-03-01
电源技术(2015年9期)2015-06-05
中国洗涤用品工业(2015年11期)2015-02-28
