
对国家食品药品监督管理总局2017年无菌医疗器械生产企业飞行检查的分析与思考
2018-03-02 03:33宋海辉马仁杰许游游金术董宇琼
中国医疗器械信息 2018年1期
宋海辉 马仁杰 许游游 金术 董宇琼
杭州艾力康医药科技有限公司 (浙江 临安 311305)
国家食品药品监督管理总局于2014年12月29日发布《医疗器械生产质量管理规范》(2014年第64号)[1],办法规定《医疗器械生产质量管理规范》自2015年3月1日起施行,同时国家食品药品监督管理总局发布了《关于医疗器械生产质量管理规范执行有关事宜的通告》(2014年第15号),通告规定自2016年1月1日起,所有第三类医疗器械生产企业应当符合医疗器械生产质量管理规范的要求[2]。同年国家食品药品监督管理总局于2015年09月01日起实施《药品医疗器械飞行检查办法》[3],并于2015年09月25日发布《食品药品监管总局关于印发医疗器械生产质量管理规范现场检查指导原则等4个指导原则的通知》(食药监械监[2015]218号)[4],自此为医疗器械生产企业质量管理体系检查奠定了基础。
1.检查基本情况
截止至2017年07月30日,2017年度国家食品药品监督管理总局对医疗器械生产企业共执行飞行检查45次,被查企业为无菌医疗器械生产企业有25家,诊断试剂生产企业6家,植入医疗器械生产企业7家,处于停工的企业有7家,有效检查企业为38家。检查结论为:停产整改11家(含10家无菌医疗器械生产企业和1家植入医疗器械生产企业),限期整改27家,被要求停产整改企业占所有开工企业的比例高达28.95%,而无菌生产企业中停产的比例更是高达40.00%。
2.缺陷项目分布情况
2.1 缺陷项目在企业中分布概况
无菌医疗器械生产企业被飞行检查25次,总缺陷项为318条。被要求停产整改企业10家,严重缺陷项有42条,一般缺陷项107条,平均每家企业占14.9条。被要求限期整改的企业一般缺陷项共169条,平均每家企业占11.27条,被要求停产的企业缺陷项比要求限期整改的企业缺陷项相对要多。
2.2 被要求停产行业分布情况
在被检查的25家无菌医疗器械生产企业中,其中有10家被要求停产整改,10家被要求停产的生产企业主要分布在软性亲水接触镜行业占40%和一次性输液器(针)行业占30%。今年被要求停产概率之高,应当引起全国所有无菌医疗器械生产企业的重视。
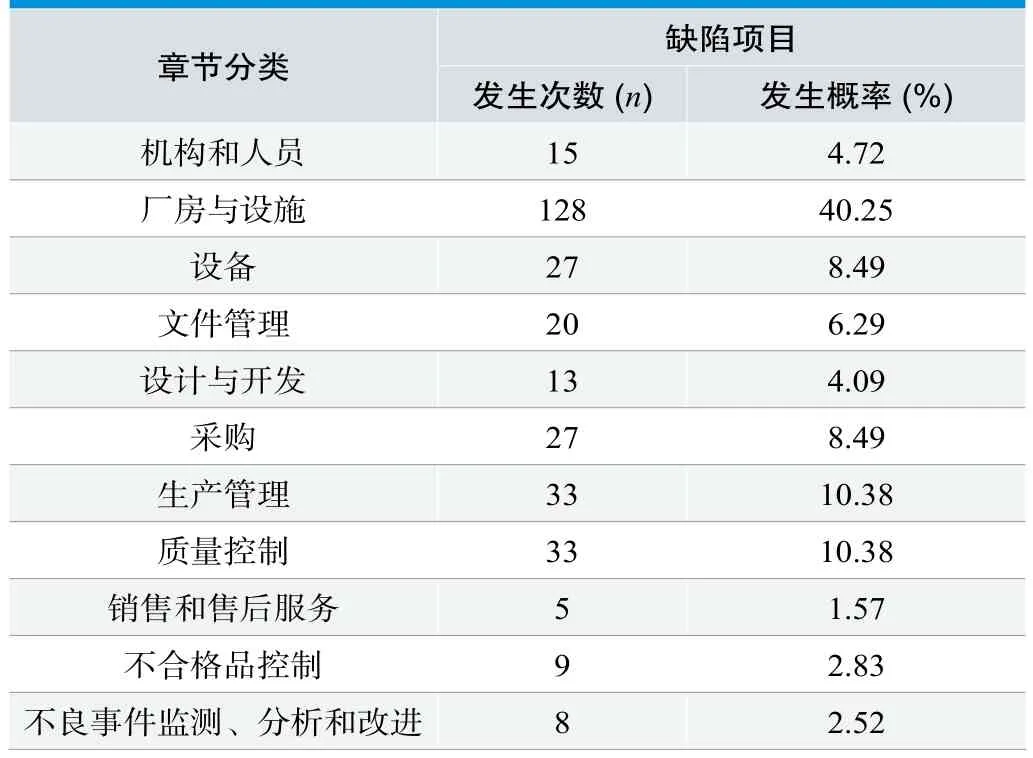
表1. 缺陷项目在《医疗器械生产质量管理规范》及其无菌检查指导原则的分布情况
2.3 缺陷项目在标准条款中的分布情况
将25家被检查到的无菌医疗器械生产企业所发生的缺陷项在《医疗器械生产质量管理规范》及其无菌检查指导原则中的分布进行简单统计,统计数据见表1。
发生概率较高的项目说明其在实施过程中是比较容易出现问题的项目,从表1中得出,厂房与设施、生产管理、质量管理是比较容易出现缺陷项的情况。
2.4 严重缺陷项分布情况
10家无菌医疗器械企业因存在严重缺陷项而被要求停产整改,共涉及到42条严重缺陷项,企业存在严重缺陷项条目最少的为1条,最多的条目有9条,严重缺陷项条目较多的企业其一般缺陷项条目也较多,42条严重缺陷项在《医疗器械生产质量管理规范》及其指导原则中的分布情况分布为厂房设施占19.05%,采购占16.67%,生产管理占19.05%,质量控制占21.43%,其他项占23.80%。
3.讨论
3.1 无菌医疗器械生产企业如何应对飞行检查
无菌医疗器械生产企业对飞行检查应该并不陌生,2009年国家食品药品监督管理总局就发布过《医疗器械生产质量管理规范(试行)》规定,要求三类的医疗器械应该执行此规范,所以《医疗器械生产质量管理规范》并不是无中生有的东西,执行这些规范是有一定的前期基础,但从3.1章可以看出,情况并不是非常乐观,这可能与企业缺乏对《医疗器械生产质量管理规范》的正确理解与认识有关[5]。
3.2 厂房设施维护保养不到位
厂房设施维护保养不到位在表1中表现的非常抢眼,很多企业对规范中附录部分的厂房与设施没有关注到位,导致很多企业不符合附录中有关厂房与设施的章节。特别是微生物限度和无菌检测室,是很多企业容易遗忘的管理区域;还有一部分对厂房的管理不到位,生产区容易出现“跑、冒、滴、漏”现象,生产区设备比较陈旧,表面出现脱落、锈迹的现象,容易对产品造成污染;工艺用水的管理也应该引起足够的重视,如果工艺用水不符合要求,势必对产品也造成一定程度的影响;工艺用气特别与内包装直接接触的工艺用气,有些企业没有对工艺用气进行验证,以证明工艺用气不会对产品质量造成影响。总之,厂房设施关系到方方面面,企业在日常巡查或者内审时发现有不合理的地方,就应该及时采取预防纠正措施,避免给企业带来不必要的损失。
3.3 采购管理
采购管理也是容易出现严重缺陷项。没有与供应商签订质量协议,采购的产品不符合国家或行业标准,没有制定有效的采购控制程序,采购的物资没有按照要求进行验证,采购物资无法满足追溯,这些都有可能会造成严重缺陷项。同时,在满足规范的要求前提下,企业还应该关注《医疗器械生产企业供应商审核指南》,指南中明确要求对提供灭菌服务的分包商应该进行现场审核,对采购的物资有洁净要求的应该对供应商进行现场审核,以资料证明其能够满足物料的洁净度要求。
3.4 生产管理和质量管理
生产管理与质量管理是企业生产活动的主要内容。企业生产记录不完善,生产记录不能满足可追溯的要求,生产记录物料平衡相差较多,无法解释去向,产品放行早于成品检验结束时间,产品检验缺项或者少检,出厂检验报告没有涵盖国家标准或者行业标准,无批准放行记录,这些都可能会成为严重缺陷项。企业应该关注《医疗器械生产企业质量控制与成品放行指南》的内容,有必要将其内容转化到企业程序文件或者三级管理文件中,确保产品质量符合产品技术要求或者国家强制标准的要求,避免上市后的风险。
4.小结
本文仅对国家食品药品监督管理总局2017年医疗器械生产企业飞行检查数据进行了简单分析,总体来说,检查现状不容乐观,如何从容应对飞行检查,这是企业所应该思考的问题,也应该着手进行解决的问题,毕竟被要求停产整改,就意味着整个企业陷入了水深火热之中,还应该从人、机、料、法、环各方面入手,查漏补缺,保证质量管理体系持续,高效运行。
[1]国家食品药品监督管理总局.医疗器械生产质量管理规范[S].国家食品药品监督管理总局公告2014年第64号.
[2]国家食品药品监督管理总局.关于医疗器械生产质量管理规范执行有关事宜的通告[Z].国家食品药品监督管理总局通告2014年第15号.
[3]国家食品药品监督管理总局.药品医疗器械飞行检查办法[Z].国家食品药品监督管理总局令第14号.
[4]国家食品药品监督管理总局.食品药品监管总局关于印发医疗器械生产质量管理规范现场检查指导原则等4个指导原则的通知[Z].食药监械监[2015]218号.
[5]张永秉,梁毅,丁静.医疗器械生产企业在GMP实施中采购管理的常见问题及对策研究[J].中国医疗器械信息,2017,23(8):3-6.
猜你喜欢
现代仪器与医疗(2022年1期)2022-04-19
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
生物医学工程学进展(2021年3期)2021-01-20
现代装饰(2020年7期)2020-07-27
质量安全与检验检测(2019年3期)2019-07-31
质量安全与检验检测(2018年6期)2018-12-28
消费导刊(2017年12期)2017-09-29
消费导刊(2017年13期)2017-09-15
人民周刊(2016年10期)2016-06-02
