
利用黄褐棉染色体片段导入系定位产量和纤维品质性状QTL
2018-01-22 01:10李定国聂以春林忠旭
作物学报 2017年12期
沈 超 李定国 聂以春 林忠旭,*
利用黄褐棉染色体片段导入系定位产量和纤维品质性状QTL
沈 超1李定国2聂以春1林忠旭1,*
1华中农业大学植物科学技术学院作物遗传改良国家重点实验室, 湖北武汉 430070;2长江大学农学院, 湖北荆州 434025
陆地棉的遗传基础狭窄, 阻碍了棉花的遗传改良进程。为有效拓宽陆地棉的遗传基础, 本试验利用野生种黄褐棉(AD4)为供体亲本, 以综合性状优良的B0011品系为受体亲本(国审棉华杂棉H318的亲本之一), 构建了含71个株系的导入系BC5S5群体。基于SLAF-seq的基因分型和多年多点田间试验的综合分析表明, 该导入系在产量和纤维性状方面具有很大的变异, 共检测到48个QTL, 其中包含19个产量和29个纤维构成因素相关的QTL。在At亚组检测到9个性状的32个QTL, 在Dt亚组为16个。进一步对QTL加性效应方向分析显示, 其中有30个QTL的加性效应为正, 18个QTL的加性效应为负。本研究结果为利用黄褐棉重要农艺性状有利等位基因改良陆地棉产量和品质奠定了基础。
陆地棉; 黄褐棉; 导入系; 产量; 纤维品质; QTL; 加性效应
异源四倍体棉花有5个种, 栽培种主要有陆地棉和海岛棉, 其余3个为野生种[1], 其中陆地棉的产量占世界棉花产量的95%以上[2]。然而, 陆地棉的遗传多样性低, 且遗传基础狭窄, 这成为陆地棉改良的主要限制因素[3]。因此, 利用野生棉资源对拓宽陆地棉的遗传多样性具有重要的潜在育种价值。
棉花的产量和纤维品质性状是复杂的数量性状,受多基因控制[4]。它们的表型受基因与环境共同调控, 且它们之间往往存在显著负相关[5]。通常, QTL定位是基于F2/F2:3、BC1、DH、RIL等分离群体[6-7]; 然而, 这些群体的遗传背景比较复杂, 加之数量性状本身的复杂性, 且QTL定位还易受非目标QTL的干扰, 严重制约了对目标性状的精细解析。导入系是QTL精细定位和复杂农艺性状解析的理想材料, 能克服种间杂交分离群体对基因渐渗的影响。导入系是通过杂交、回交、分子标记辅助选择等方法构建的能够覆盖整个作物基因组的一系列近等基因系,其整个遗传背景只含有供体亲本的一个或几个染色体片段, 其它部分则与轮回亲本完全相同, 能够消除遗传背景的噪音, 预测到那些在F2或者RIL等初级群体中被掩盖的QTL[8]。目前, 在不同的作物中都已经进行了深入的研究, 例如番茄[9]、玉米[10]、水稻[11-12]、大豆[13]、大麦[14]、油菜[6,15]、棉花[4,16-21]等。
目前, 棉花导入系的构建主要利用海岛棉和陆地棉的种间杂交[4,16-21]。Wang等[16]构建了以陆地棉标准系TM-1为背景的海岛棉海7124的导入系。朱亚娟等[17]利用陆地棉标准系TM-1为背景的海岛棉海7124的导入系的2个家系IL-15-5和IL-15-5-1构建了F2和F2:3分离群体, 定位了衣分和籽指等与产量相关的QTL。王鹏等[18]同样利用陆地棉标准系TM-1为背景的海岛棉海7124的导入系检测到与光合色素相关的QTL, 解析了光合色素含量的遗传基础。付央等[19]用陆地棉标准系TM-1为背景的海岛棉3-79的家系Sub18为供体亲本, 以TM-1为受体亲本, 培育了海岛棉3-79第18染色体片段导入系,并利用这个特定的染色体片段导入系检测了12个产量与纤维品质性状相关的QTLs。最近, 王云鹏等[4]创制了以陆地棉中棉所8号为背景的海岛棉Piam 90-53的导入系。黎波涛等[20]以早熟陆地棉中棉所36为受体亲本, 海岛棉海1为供体亲本构建导入系群体, 筛选82个株系, 采用株系间随机成对杂交组配F1, 并在多个环境下分析发现, 导入系及F1的变异丰富, 部分亲本与 F1的皮棉产量与纤维品质在多个环境下得到同步提高。Si等[21]构建了一个以海岛棉Xinhai 25为背景, 导入陆地棉TM-1片段的导入系群体, 为我们提供了新的参考。
然而, 除海岛棉外, 利用棉花其他的野生资源相对较少。黄褐棉在3个异源四倍体野生棉中, 其亲缘关系距离陆地棉最远[1], 因此, 最有可能存在新的有利基因位点用于陆地棉的改良。肖松华等[22]研究显示, 黄褐棉渐渗陆地棉株系不仅对棉花黄萎病达到抗性水平, 而且在棉花纤维品质方面也存在着丰富的遗传变异。汪保华等[23]研究表明, 3个黄褐棉与陆地棉构建的近等基因系的纤维强度与细度表现十分突出。
为利用野生棉种质资源拓宽陆地棉遗传多样性, 本研究以综合性状优良的B0011品系为受体亲本, 以野生种黄褐棉为供体亲本, 构建以陆地棉为背景, 含有黄褐棉不同片段长度的导入系群体, 进行棉花产量和纤维品质相关性状的QTL定位, 希望通过黄褐棉与陆地棉之间的基因渐渗转移, 拓宽陆地棉的遗传基础, 为以后的棉花品种遗传改良奠定基础。
1 材料与方法
1.1 试验材料
轮回亲本B0011是华中农业大学选育的常规棉花品系, 其综合性状好, 是华杂棉H318的亲本之一[24]。供体亲本黄褐棉, 具有纤维品质优良, 抗黄萎病和抗虫等特性[22,25]。以B0011为母本与杂交产生F1, 以B0011为轮回亲本回交5次, 通过单籽传法, 获得71个BC5株系; 用获得的株系通过单籽传法连续自交5代, 最后得到含有71个株系的BC5S5导入系群体。
1.2 基因组DNA的制备和SLAF-seq
选取幼嫩的叶片, 用液氮速冻, 存于–70℃冰箱备用。然后, 用Plant Genomic DNA Kit (TIANGEN Biotech, Beijing, China)试剂盒提取基因组DNA, 并用NanoDrop 2000C Spectrophotometer (Thermo Scientific, USA)和琼脂糖凝胶电泳检测, 以保证满足建库要求。之后, 进行SLAF-seq的建库、测序、标签分析以及SNP鉴定[26-27]。亲本测序深度20×以上, 子代测序深度5×以上。
1.3 田间种植和性状调查
2015年在湖北省黄冈市(E1)和荆州市(E2)及2016年在湖北省荆州市(E3)和鄂州市(被洪水毁坏)种植71个导入系家系。种植地点分别为湖北省黄冈市农业科学院试验田和长江大学试验田。每年每点2次重复, 并且按照随机区组种植。单行小区每行长5.0 m, 宽0.8 m, 种植10株。人工摘取每个家系中部20朵棉花进行产量和纤维品质的考种[28]。产量性状包括单铃籽棉重(SCW)、单铃皮棉重(LW)、衣分(LP); 纤维品质性状包括: 纤维长度(FL)、整齐度(FU)、马克隆值(MIC)、伸长率(FE)、比强度(FS)、短纤维率(SF)。
1.4 数据的分析和QTL定位
用SPSS 17.0 (SPSS, Chicago, Illinois, America)软件分析群体平均数等特征数据。基于一个线性模型[29], 用最佳无偏向性预测(The best linear unbiased prediction, BLUP, http://www.eXtension.org/pages/61006)评估每个株系在这3个环境中的表型值。以Wang等[30]的遗传图谱为参照, 将标记序列用BLASTN的方法比对到参考基因组[31], 阈值为E≤1e−5, 选取最佳匹配且与遗传图谱顺序一致的标记。之后, 将在2个标记之间大于等于3个SNP的片段定义为导入片段, 并分析其在各染色体及基因组的分布情况。
由于本研究群体是非标准单片段导入系, 故不能用以往的检验来定位QTL。然而, Wang等[32-33]提出了一种可以检测多片段导入系QTL方法。它是基于逐步回归的极大似然估计算法(RSTEP-LRT), 采用逐步回归分析选择对目标性状最重要的片段或标记, 然后通过似然比检验来计算每个染色体片段或标记的LOD值。对于理想的导入系而言, 它在统计学上与标准的检验是等效的。利用QTL IciMapping 4.1 (http://www.isbreeding.net/)软件[32]检测产量和纤维品质相关的加性效应QTL。根据QTL IciMapping 4.1[33]软件, 对SNP进行基因分型, 将与受体亲本B0011相同的基因型记为“0”, 与供体亲本黄褐棉相同的基因型记为“2”, 进行QTL定位。以LOD≥3为阈值判断其是否存在加性效应的QTL。参照McCouch等[34]的规则命名QTL。参考Wang等[35]的染色体示意图(着丝粒区域)。
2 结果与分析
2.1 亲本和群体的产量和纤维品质性状在多个环境中的表现
根据偏度绝对值小于1, 认为该性状群体符合正态分布。从表1可知, 除了SCW和LW在E1, LW和LP在E2, FU在E3外, 其他所有性状均呈现连续分布, 群体符合正态分布, 并且, 各性状在不同的环境中均表现出差异。由图1可知, 产量和纤维性状在多个环境中的BLUP值均表现为相对连续的正态分布, 并且分离变异较大。
SCW: seed cotton weight per boll; LW: lint weight per boll; LP: lint percentage; MIC: micronaire; FL: fiber length; FU: fiber uniformity; SF: short fiber; FS: fiber strength; FE: fiber elongation; Env.: environment; Min.: minimum value; Max.: maximum value; Mean: mean value; SD: standard deviation.
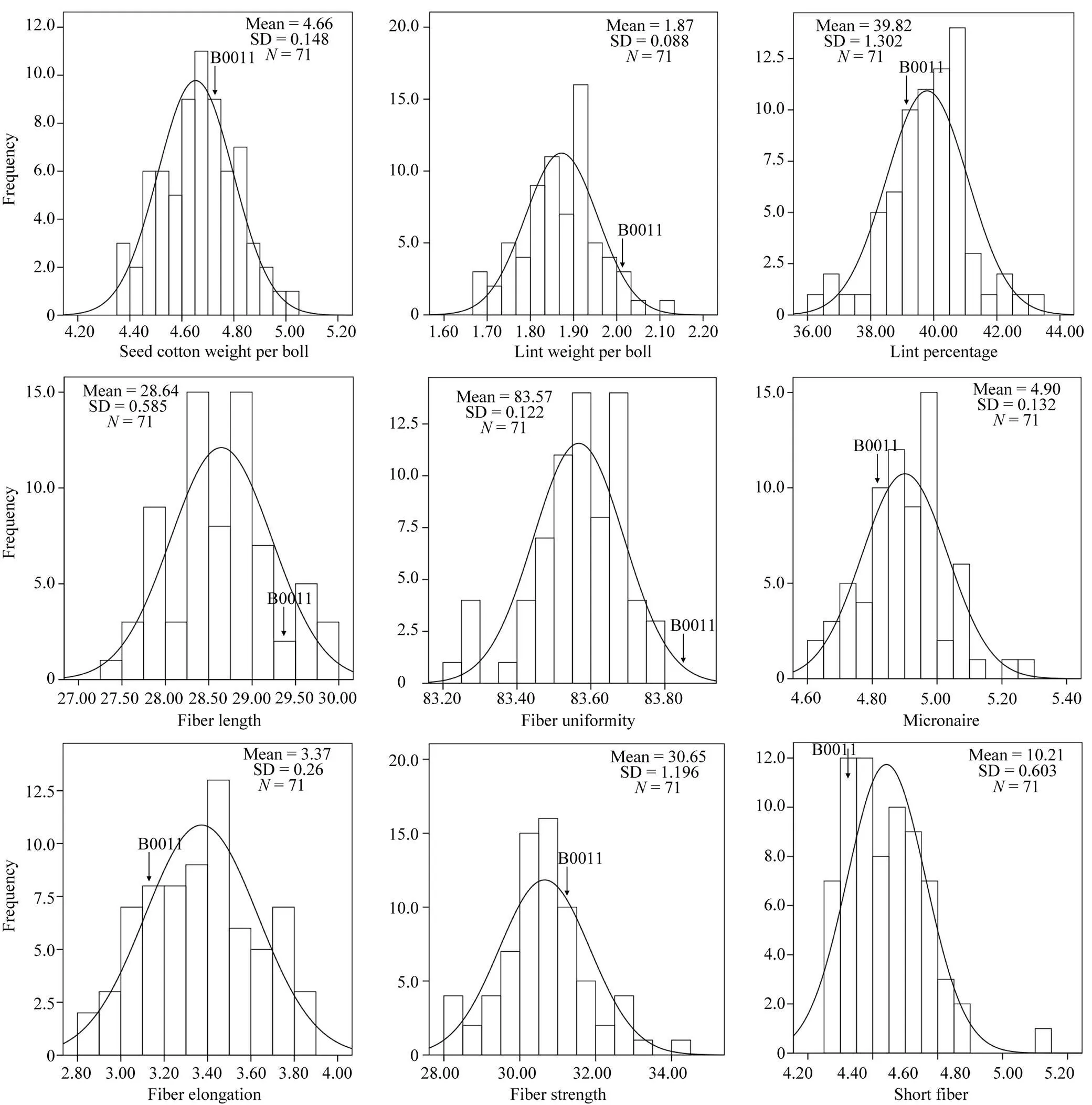
图1 产量和纤维品质性状在多个环境中BLUP的频率分布
方差分析(表2)表明, 除纤维整齐度(FU)与短纤维率(SF)的基因×环境的互作效应没有达到显著水平外, 其他性状都受到基因型、环境效应以及基因×环境的互作效应的强烈影响, 其值都达到了极显著水平, 对性状的影响为环境效应>基因型>基因×环境的互作效应, 显示性状的差异主要是由不同年份和不同种植环境造成的。
2.2 产量和纤维品质性状之间的相关性分析
从图2可知, 单铃籽棉与单铃皮棉呈正相关。单铃皮棉与衣分、马克隆值、伸长率呈正相关, 与纤维长度、比强度呈负相关。衣分与马克隆值、伸长率呈正相关, 与纤维长度、比强度呈负相关。马克隆值与比强度呈正相关; 与纤维长度呈负相关。纤维长度与纤维整齐度、比强度呈正相关, 与短纤维率、伸长率呈负相关。纤维整齐度与比强度呈正相关, 与短纤维率呈负相关。纤维比强度与短纤维率呈负相关。
2.3 基于SNP标记的导入片段分布
通过SLAF-seq测序, 在BH导入系群体中总共发现了2839个SNP, 不均匀地分布在各个染色体上, 染色体A13上含有的SNP最多, 为393个, 而染色体D03最少, 仅有19个(图3)。对于各株系而言, 也呈现不均匀分布, 其中BH2系含有的SNP最多, 为646个; 而BH65系仅有96个(图4)。对BH群体的71个株系的基因型分型发现, 在At亚组的A02、A06、A09、A12和A13染色体上及Dt亚组的D04、D06、D12和D13染色体上, 不同的家系间存在相同的导入片段, 尤其在A13和D06染色体最多(图5)。
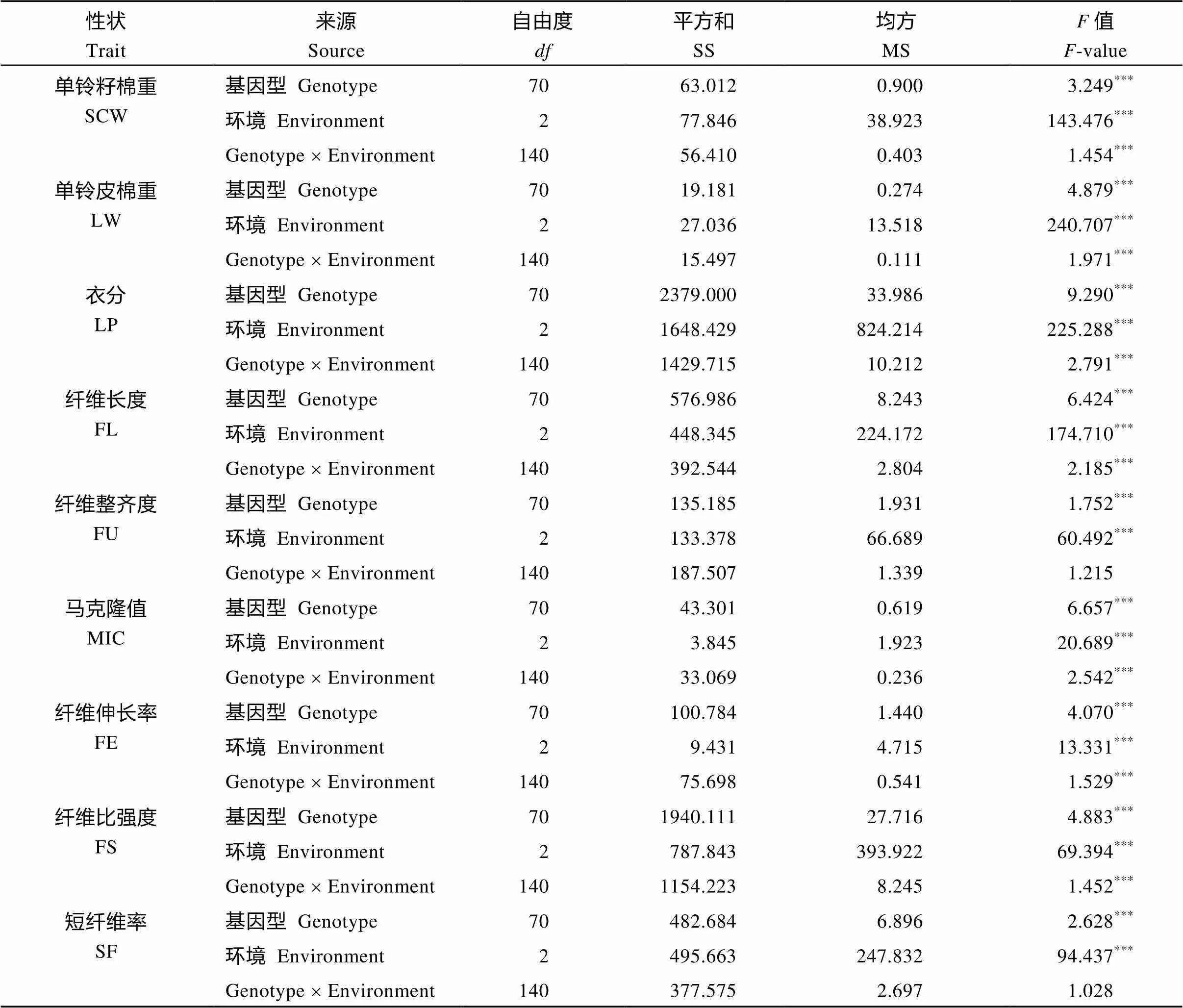
表2 产量和纤维性状的不同环境间的双向方差分析
*表示0.05水平差异显著;**表示0.01水平差异显著;***表示0.001水平差异显著。
SCW: seed cotton weight per boll; LW: lint weight per boll; LP: lint percentage; MIC: micronaire; FL: fiber length; FU: fiber uniformity; SF: short fiber; FS: fiber strength; FE: fiber elongation.*Correlation is significant at the 0.05 probability level;**Correlation is significant at the 0.01 probability level;***Correlation is significant at the 0.001 probability level.
通过将测序的SNP标记与遗传图谱比较分析, 发现在本群体中导入片段的总长度为2030.0 cM, 其中A亚组和D亚组分别为1093.7 cM和936.5 cM, 导入片段所占的比率分别是40.0%和32.7%。A11染色体的导入片段的长度最短, 为31.6 cM, 而A06染色体的导入片段长度最长, 为137.8 cM。导入片段在基因组的覆盖率从A11的11.9%到A13的99.9%。其中在A02、A06、A10、A12、A13、D07和D10染色体上的导入片段覆盖率大于50%, 而A11, D05和D09染色体的导入片段覆盖率小于20%, 整个基因组的导入片段的平均覆盖率为36.3% (表3)。
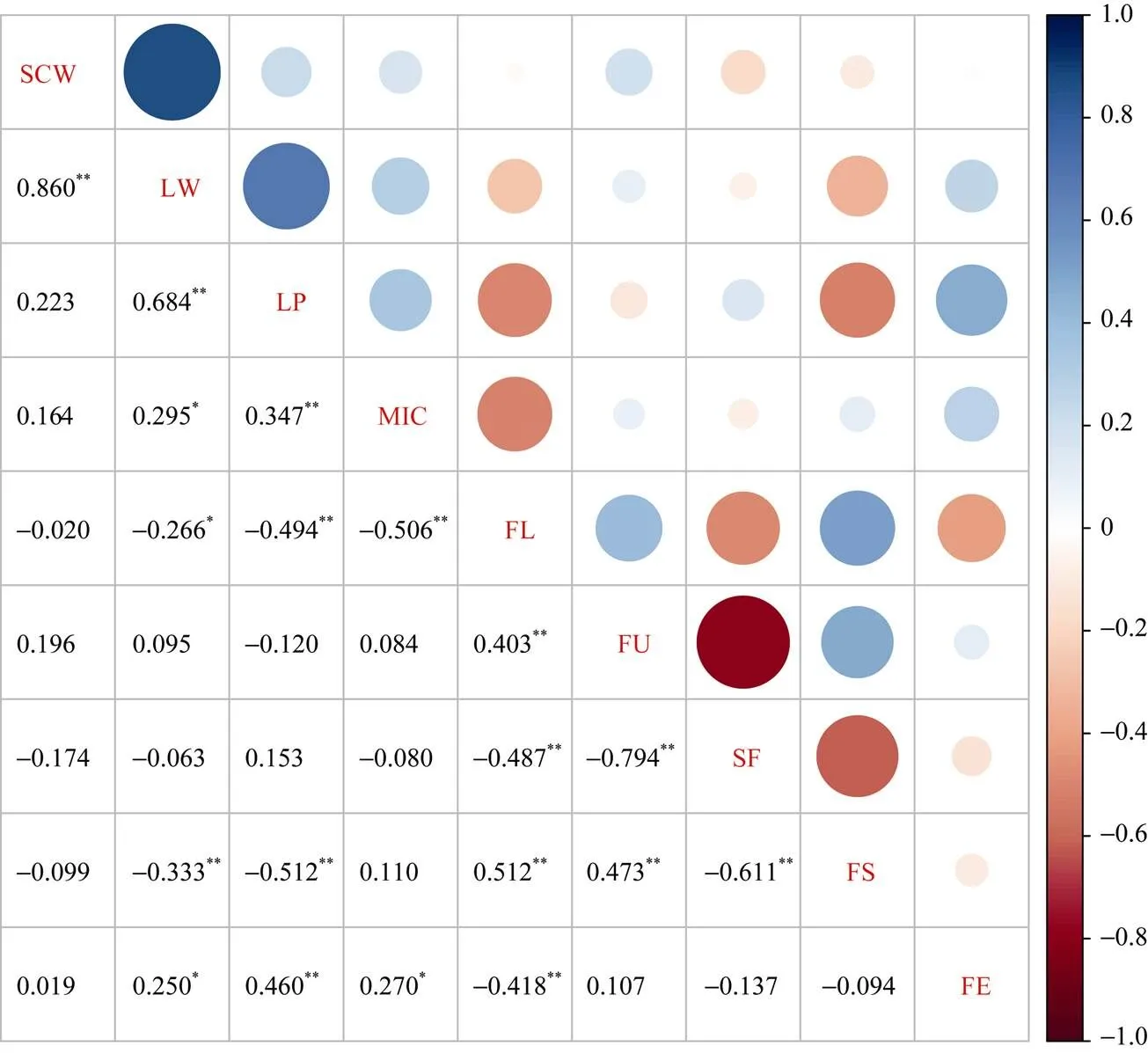
图2 产量与纤维品质性状间的Pearson’s相关系数
*表示0.05水平差异显著;**表示0.01水平差异显著。
*Correlation is significant at the 0.05 probability level;**Correlation is significant at the 0.01 probability level. SCW: seed cotton weight per boll; LW: lint weight per boll; LP: lint percentage; MIC: micronaire; FL: fiber length; FU: fiber uniformity; SF: short fiber; FS: fiber strength; FE: fiber elongation.
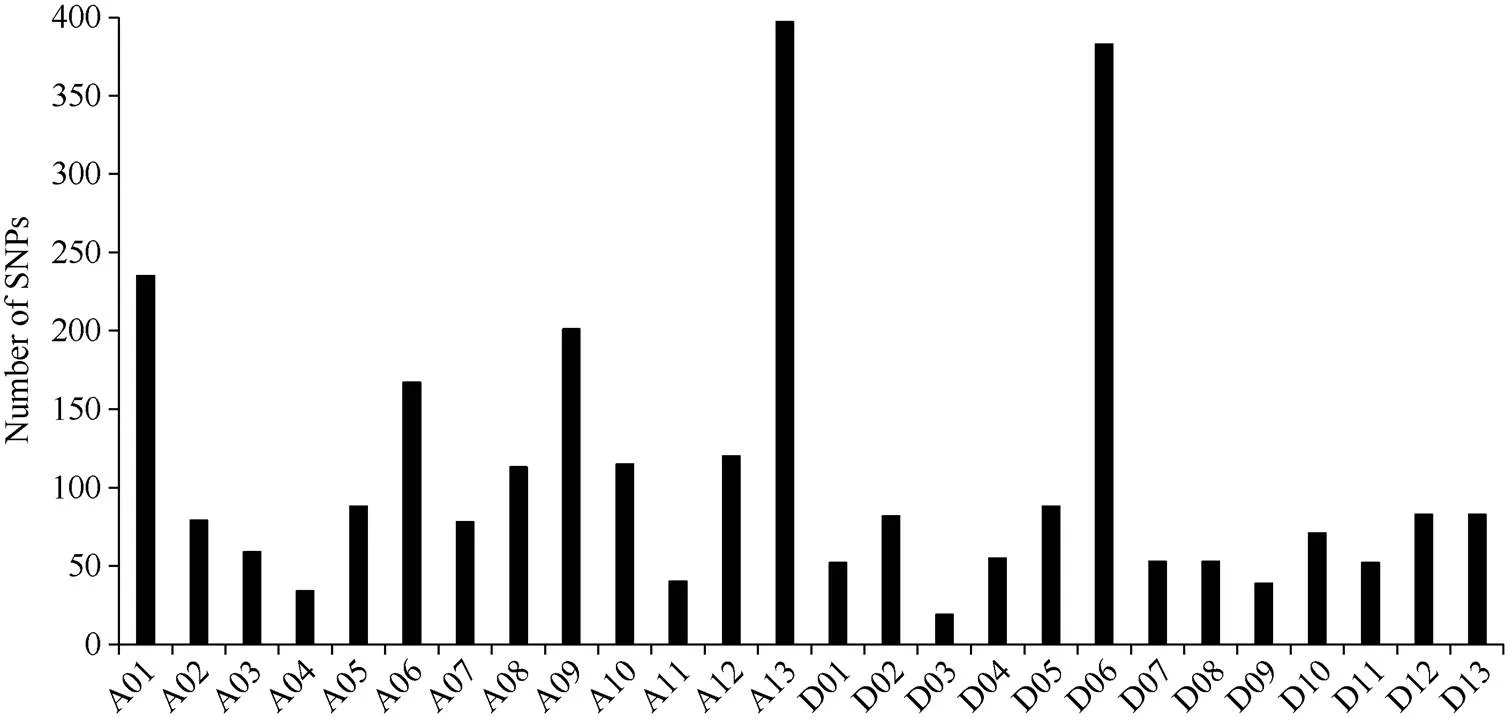
图3 SNP在每条染色体上的的分布
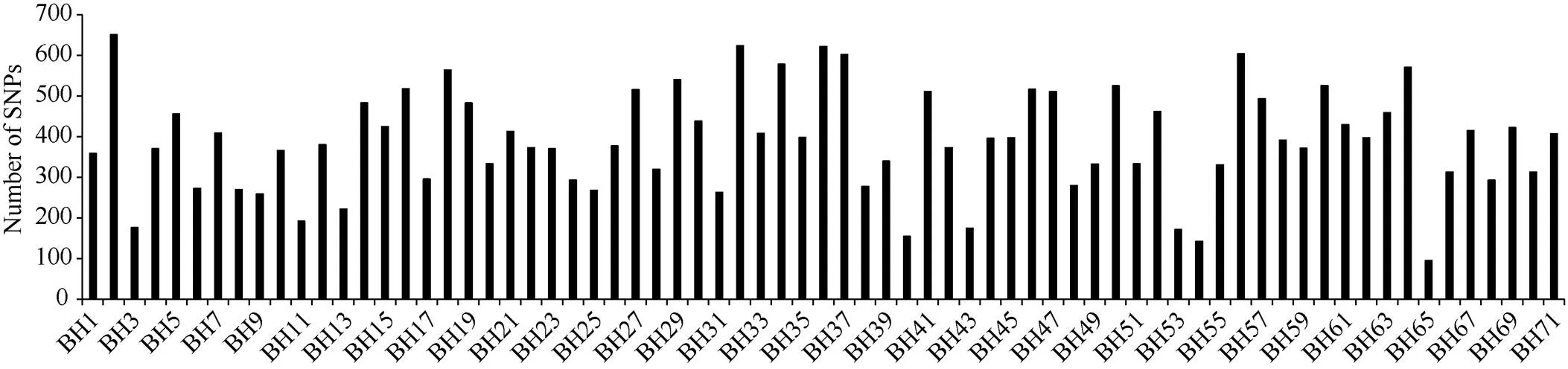
图4 SNP在71个株系中的分布
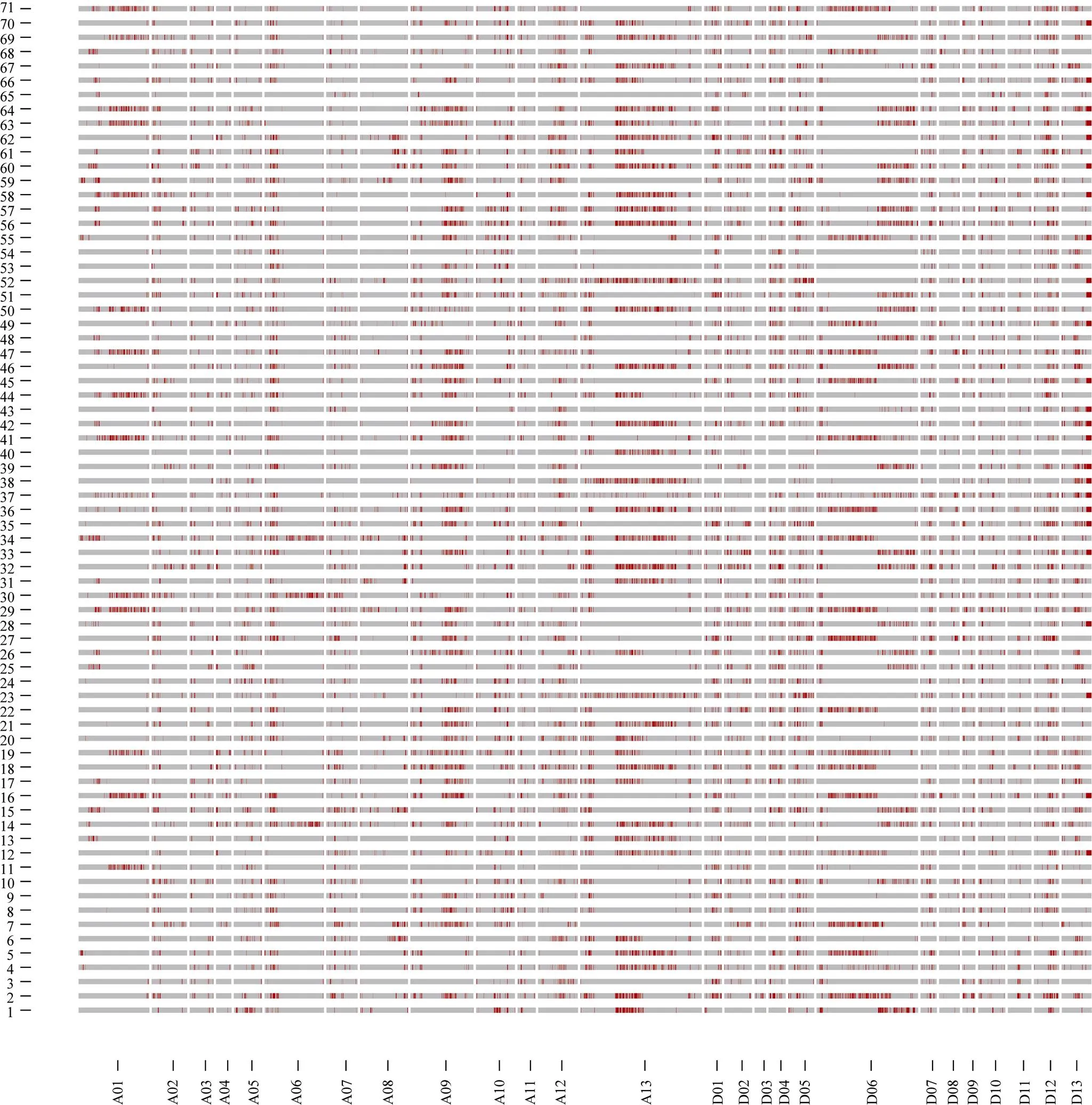
图5 71个株系的图示基因型(灰色为B0011, 红色为黄褐棉)
2.4 产量和纤维品质相关性状的加性QTL定位
用QTL IciMapping 4.1软件总共检测到48个QTL, 分布在除A04、A11、D01、D06和D13以外的21条染色体上, 在A08染色体上检测到的QTL最多, 均为6个; 其次是在A05、A09、D05为5个, 而在A03、A06、D02、D03、D04、D07、D08、D09、D11和D12染色体上检测到的QTL最少, 均为1个, 这其中包含19个与产量和29个与纤维品质相关的QTL (表4、表5和图6)。没有检测到纤维长度(FL) QTL。在A05染色体上的22.06 Mb位点同时检测到分别与单铃籽棉()和单铃皮棉()相关的2个QTL, 在35.08 Mb位点也同时检测到分别与单铃籽棉()和单铃皮棉()相关的2个QTL (图6-A)。
2.4.1 产量相关性状QTL 对单铃籽棉重(SCW)来说, 共检测到3个QTL, 分布在2条染色体上(A01, A05), 解释表型变异11.70%~31.00%, LOD值为3.06~5.63, 加性效应值为–0.46~1.45 (表4、图6-A和图7-A); 而对单铃皮棉重(LW), 检测到4个位点, 主要分布在3条染色体上(A05、A09和D06), 解释表型变异13.85%~29.10%, LOD值为3.21~5.00, 加性效应值为–0.30~0.53 (表4、图6-A和图7-A); 对衣分(LP), 则检测到12个QTL, 主要分布在10条染色体上(A01、A05、A06、A08、A09、A10、A13、D03、D04和D06), 解释表型变异6.70%~28.04%, LOD值为3.05~7.66, 加性效应值为–3.33~3.29 (表4、图6-A和图7-A)。
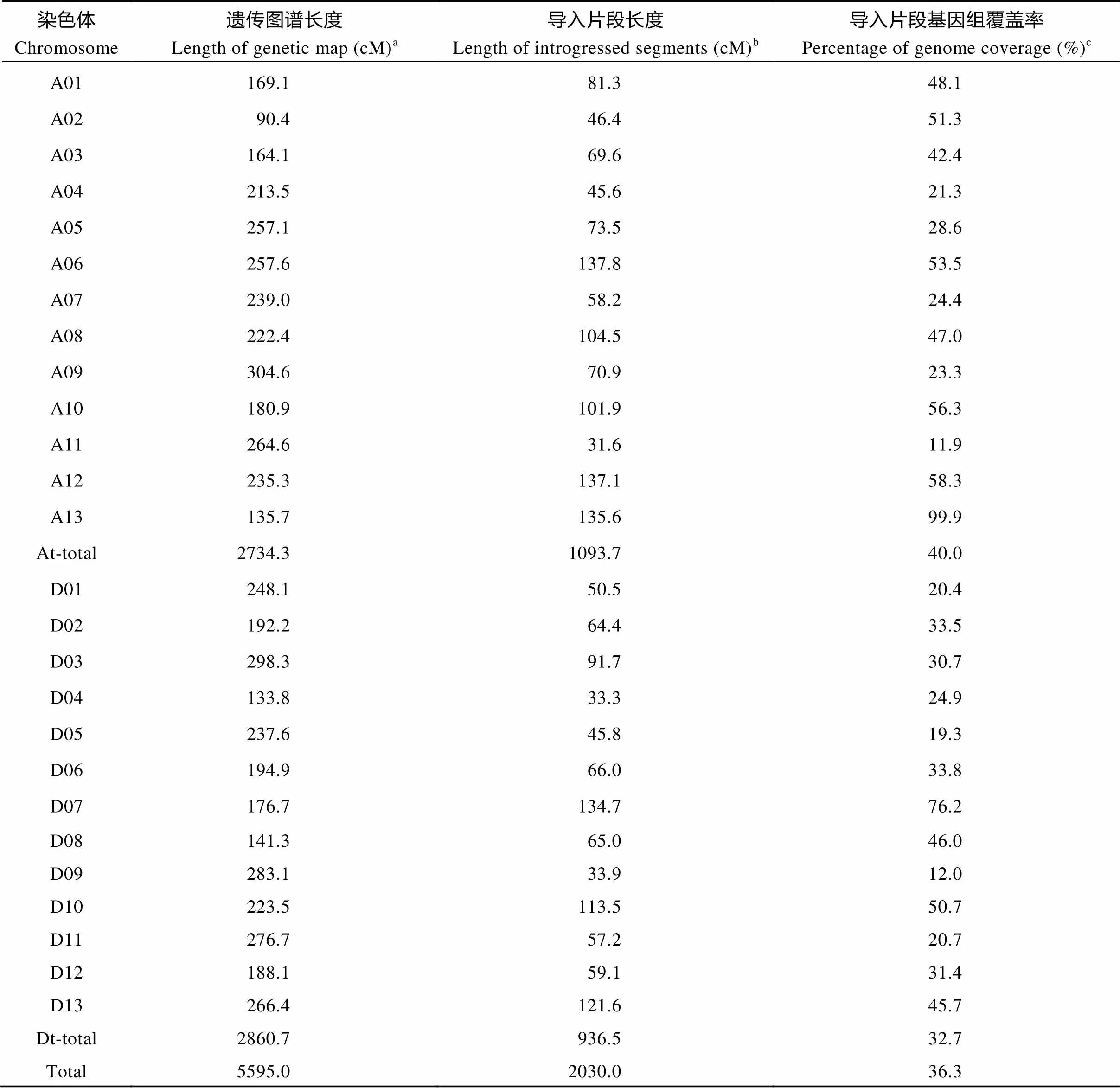
表3 导入系群体中导入片段的基因组覆盖率
aThe length of genetic distance in Wang et al.[30]bThe sum of length of introgressed segments distributed on the chromosome.cPercentage of genome coverage = c/a × 100%.
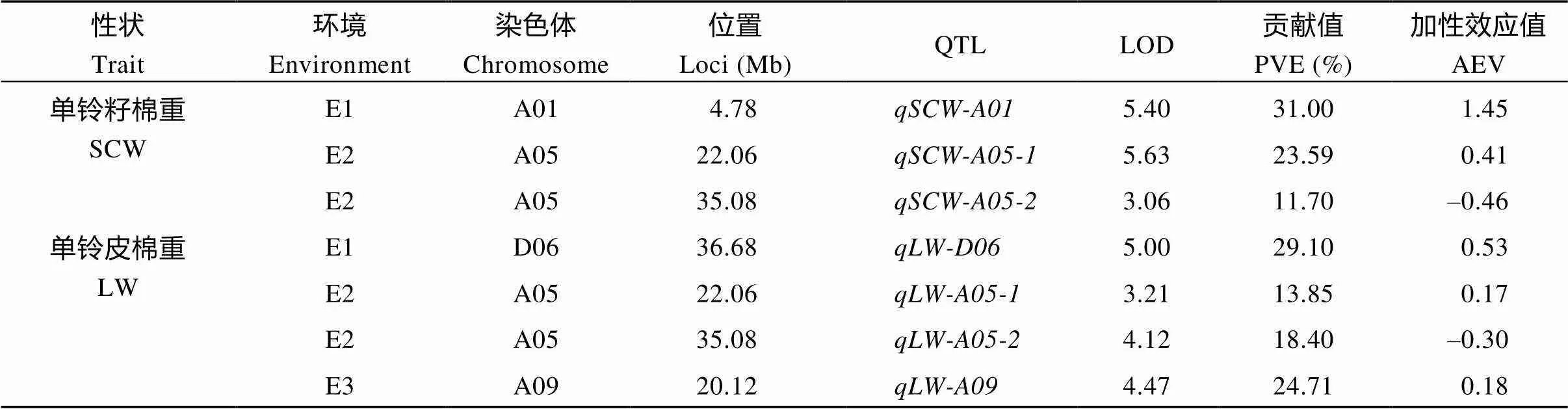
表4 产量性状的QTL相关信息
(续表4)
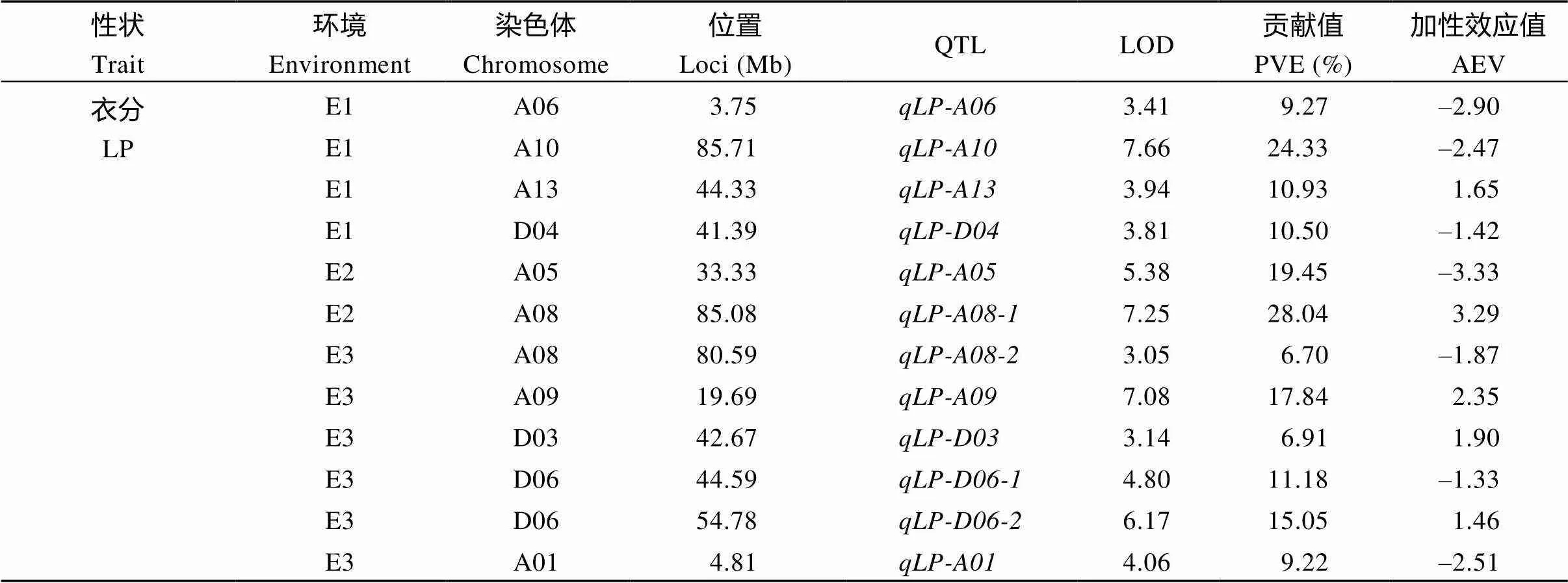
性状Trait环境Environment染色体Chromosome位置Loci (Mb)QTLLOD贡献值PVE (%)加性效应值AEV 衣分 LPE1A063.75qLP-A063.419.27–2.90 E1A1085.71qLP-A107.6624.33–2.47 E1A1344.33qLP-A133.9410.931.65 E1D0441.39qLP-D043.8110.50–1.42 E2A0533.33qLP-A055.3819.45–3.33 E2A0885.08qLP-A08-17.2528.043.29 E3A0880.59qLP-A08-23.056.70–1.87 E3A0919.69qLP-A097.0817.842.35 E3D0342.67qLP-D033.146.911.90 E3D0644.59qLP-D06-14.8011.18–1.33 E3D0654.78qLP-D06-26.1715.051.46 E3A014.81qLP-A014.069.22–2.51
SCW: seed cotton weight per boll; LW: lint weight per boll; LP: lint percentage; PVE: phenotypic variation explained by a single QTL; AEV: additive effect value.
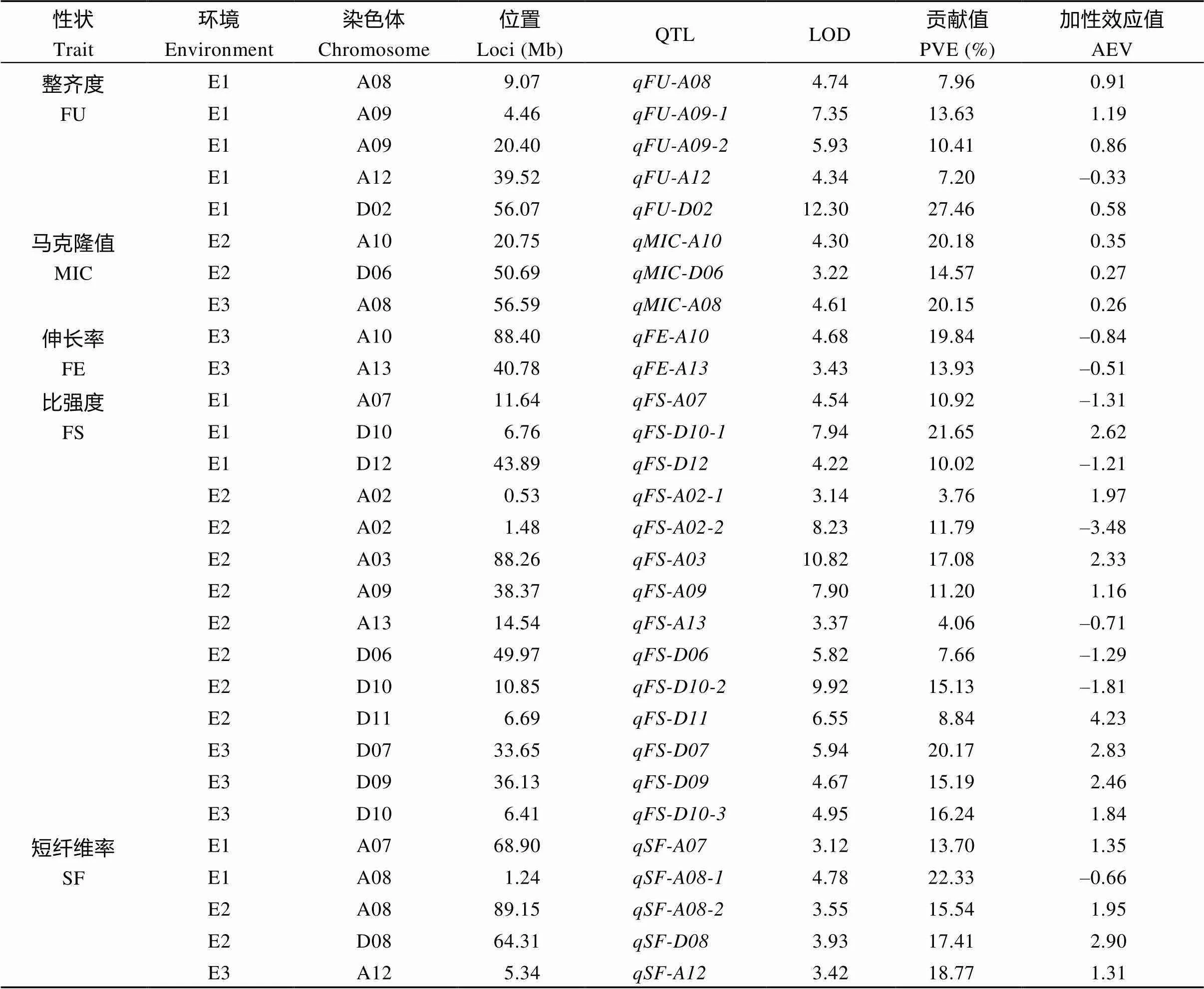
表5 纤维性状的QTL相关信息
FU: fiber uniformity; MIC: micronaire; FE: fiber elongation; FS: fiber strength; SF: short fiber; PVE: phenotypic variation explained by a single QTL; AEV: additive effect value.
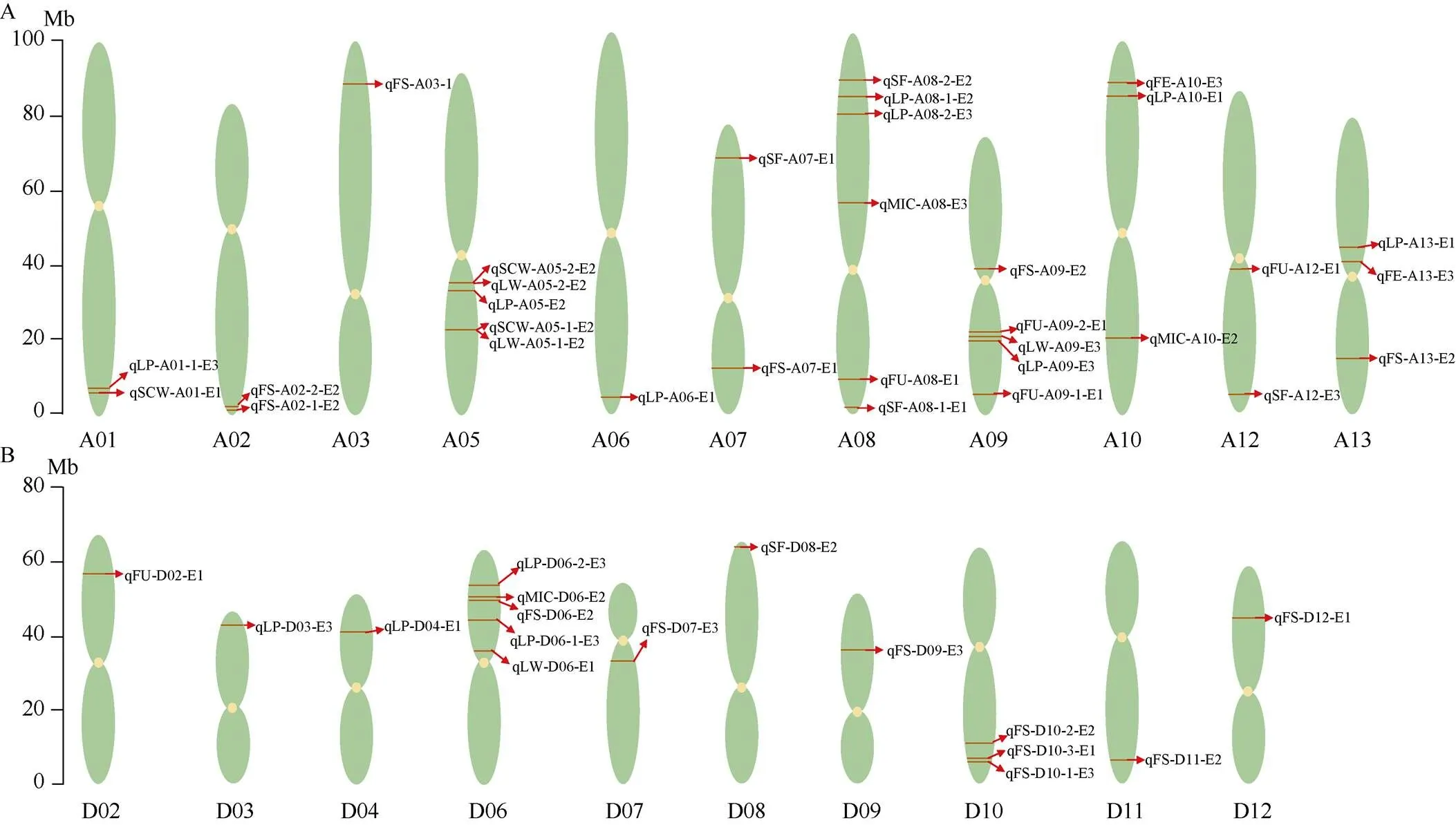
图6 产量和纤维品质的QTL在染色体上的分布示意图
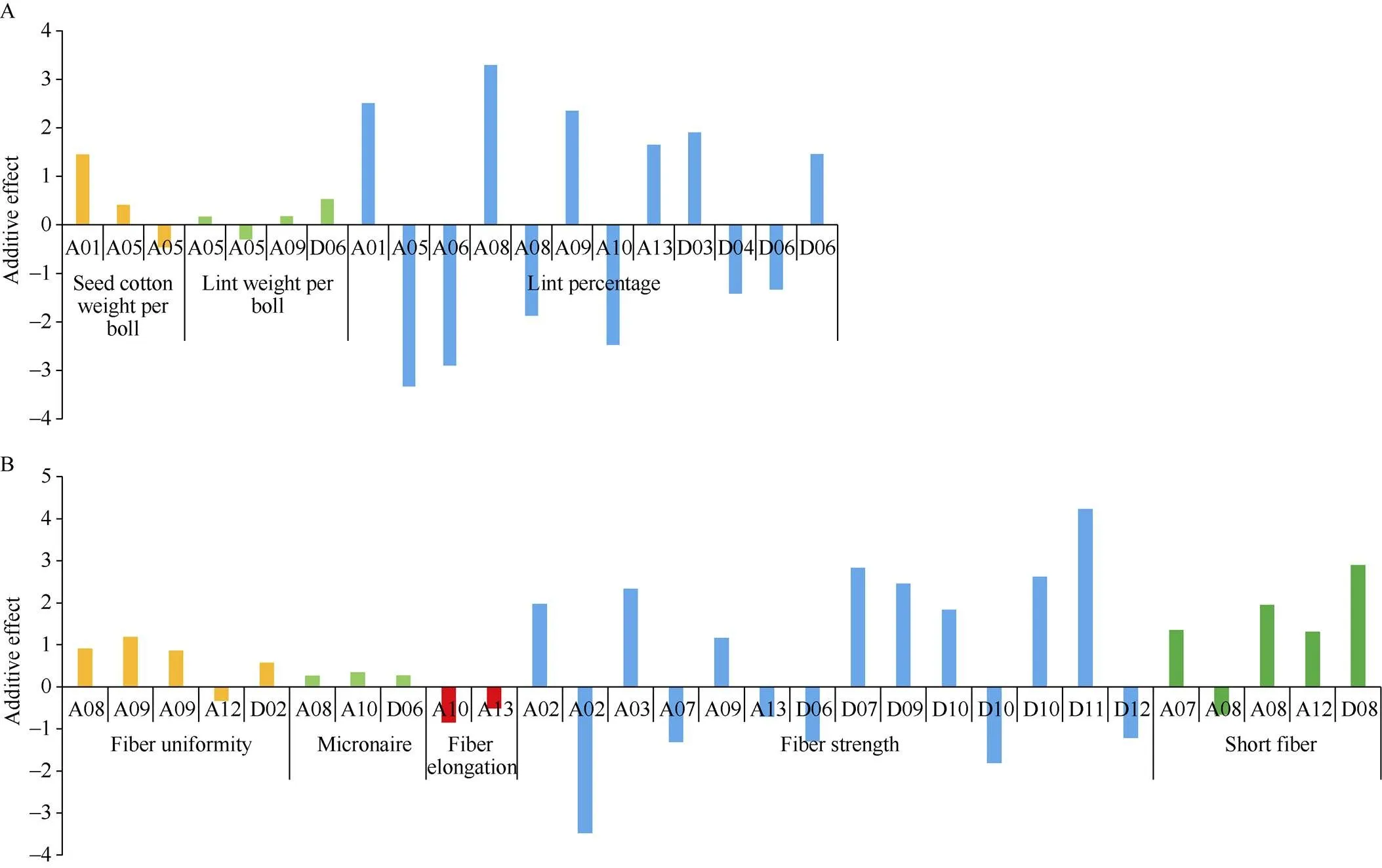
图7 产量和纤维品质的QTL在染色体上的加性效应
2.4.2 纤维品质相关性状QTL 检测到5个纤维整齐度(FU) QTL, 分布在4条染色体上(A08、A09、A12、D02), 解释表型变异7.20%~27.46%, LOD值4.34~12.30, 加性效应值为–0.33~1.19 (表5、图6和图7-B)。检测到3个马克隆值(MIC) QTL, 分布在3条染色体上(A08、A10、D06), 解释表型变异14.57%~20.18%, LOD值3.22~4.61, 加性效应值为0.26~0.35 (表5、图6和图7-B)。检测到2个纤维伸长率(FE) QTL, 分布在2条染色体上(A10、A13), 解释表型变异13.93%~19.84%, LOD值3.43~4.68, 加性效应值为–0.84~ –0.51 (表5、图6-A和图7-B)。检测到14个纤维比强度(FS) QTL, 分布在11条染色体上(A02、A03、A07、A09、A13、D06、D07、D09、D10、D11和D12), 解释表型变异3.76%~21.65%, LOD值3.14~10.82, 加性效应值为–3.48~4.23 (表5、图6和图7-B)。检测到5个短纤维率(SF) QTL, 分布在4条染色体上(A07、A08、A12、D08), 解释表型变异13.70%~22.33%, LOD值3.12~4.78, 加性效应值为–0.66~2.90 (表5、图6和图7-B)。
3 讨论
关于棉花农艺性状、品质性状、抗性性状的QTL定位已经有许多研究, 但是, 在育种中很少能够被用来进行品种的遗传改良。一个重要的原因是几乎所有的QTL都是在早期的分离群体被检测到, 使有利QTL的导入变得复杂[6]。而导入系则为QTL定位提供了一个很好的平台。目前, 在棉花中已经构建了一些导入系群体被用于QTL分析[4,16-21]。然而, 这些研究都是基于海岛棉和陆地棉构建导入系群体。对于野生棉, 尤其是黄褐棉的研究利用的报道较少。
黄褐棉原产于巴西, 在5个四倍体棉种中亲缘关系距陆地棉最远, 且具有纤维品质优良, 综合抗性好等特点[1,22,25], 因此, 引入黄褐棉优质的抗病基因将会对陆地棉的遗传改良产生极为重要的现实意义。这将会有助于提高陆地棉纤维品质及黄萎病的抗性, 从而提高陆地棉的产量及其综合特性。
通过对导入系BH群体进行SLAF-seq[26], 并过滤掉不在染色体上的SNP, 得到2839个SNPs, 发现SNPs在染色体和家系间均呈现不均匀的分布。通过对BH群体导入片段分析可以看出, 在A13和D06染色体上不同家系间大都导入了相同的片段, 这可能与其对环境的综合适应能力有关。此外, 在分析BC5家系的供体亲本基因型占全基因组的比例中发现, 最大的为3.47%, 最小的为0, 平均为0.51%, 明显低于理论比例值3.13%。与王鹏等[36]的研究相比略低, 可能是我们以遗传图谱为参照时仅选取最佳匹配且与遗传图谱顺序一致的标记, 致使图谱上标记数目减少, 而SNP再与这些最佳的标记相比即导致渗入比率偏低。
本研究对黄褐棉和陆地棉染色体片段导入系群体的3个产量和6个纤维品质性状进行了遗传分析。通过对各性状多个环境的BLUP值分析来看, 各性状均为相对连续的正态分布, 且分离变异程度较大。进一步通过对各性状多个环境的方差分析发现, 除了纤维整齐度和短纤维率没有显著地受到基因型与环境互作影响外, 其他所有性状都受到基因型, 环境, 基因型与环境互作三者极显著的影响。环境的影响效应大于环境与基因型的互作, 说明环境的差异是导致表型变异的主要来源, 也进一步说明环境效应在QTL定位中起着极其重要的作用。通过QTL定位我们发现, 在相同位点检测到与不同性状相关的QTL, 这可能是导致QTL聚集与连锁累赘的原因[28]。
已有研究显示[5,7,37-38], 相对于At亚组, QTL多在Dt亚组富集。然而, 通过对本研究检测到的QTL分析发现, 与产量性状相关的QTL, 在At亚组分布的数目比Dt亚组上多, 在At亚组上A05染色体上QTL数目较多, 在Dt亚组上D06染色体上的QTL数目最多; 而与纤维性状相关的QTL, 在At亚组上分布的数量多于Dt亚组, 在At亚组上A08染色体上QTL数目较多, 在Dt亚组上D10染色体上的QTL数目较多。最近, Si等[20]对海岛棉为背景导入系定位的39个QTL分析发现, 在At亚组上比Dt亚组具有更多QTL。说明不同的群体, 不同的标记方法, 不同的背景可能导致QTL在2个亚组间的分布不同。通过分别与Si等[20]和Wang等[30]的定位的QTL比较发现, 在A10染色体都定位到关于纤维伸长率的QTL, 然而其物理位置不同。Si等[20]定位的QTL为(NAU5323, 97.86 Mb), Wang等[30]定位的QTL为(JESPR6-BNL1161, 67.02~75.68 Mb), 我们定位的QTL为(88.40 Mb), 说明A10染色体这段区间对纤维的伸长率有很大的贡献。而在A09上(20.12 Mb)和(19.69 Mb)与Zhang等[39]定位铃重, 衣分相关QTL的位置(Gh27, 20.23 Mb)很近。对QTL加性效应方向分析发现, 在产量性状方面, 有11个QTL的加性效应方向为正, 有8个QTL的加性效应方向为负; 在纤维性状方面, 有19个QTL的加性效应方向为正, 有10个QTL的加性效应方向为负, 这表明, 黄褐棉不同的导入片段产生的效应不同。进一步利用“CSIL+F2”方法, 将更容易精细定位和克隆[17,40]这些含有具体性状特征的QTL。
4 结论
构建了含有71家系的以陆地棉为背景的黄褐棉染色体片段导入系群体, 克服了黄褐棉与陆地棉种间杂交不育, 连锁累赘等问题, 实现了将黄褐棉的优异基因引入到陆地棉的育种目标。本研究定位的产量和纤维性状相关的48个QTL将为棉花的育种提供重要的基础材料。
[1] Grover C E, Gallagher J P, Jareczek J J, Page J T, Udall J A, Gore M A, Wendel J F. Re-evaluating the phylogeny of allopolyploidL.,2015, 92: 45–52
[2] Fang D D, Jenkins J N, Deng D D, McCarty J C, Li P, Wu J. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (L.).,2014, 15: 397
[3] Zhang J F, Wu M, Yu J W, Li X L, Pei W F. Breeding Potential of introgression lines developed from interspecific crossing between upland cotton () and: heterosis, combining ability and genetic effects., 2016, 11: e0143646
[4] 王云鹏, 王省芬, 李志坤, 杨鑫雷, 张艳, 吴立强, 吴金华, 张桂寅, 马峙英. 陆地棉背景的Pima棉染色体片段置换系创制. 植物遗传资源学报, 2016, 17: 114–119 Wang Y P, Wang X F, Li Z K, Yang X L, Zhang Y, Wu L Q, Wu J H, Zhang G Y, Ma Z Y. Development of Pima cotton chromosome segment substitution lines withbackground., 2016, 17: 114–119 (in Chinese with English abstract)
[5] Jiang C X, Wright R J, El-Zik K, Paterson A H. Polyploid formation created unique avenues for response to selection in(cotton)., 1998, 95: 4419–4424
[6] Li X, Wang W, Wang Z, Li K, Lim Y P, Piao Z. Construction of chromosome segment substitution lines enables QTL mapping for flowering and morphological traits in., 2015, 6: 432
[7] Wang P, Zhu Y J, Song X L, Cao Z B, Ding Y Z, Liu B L, Zhu X F, Wang S, Guo W Z, Zhang T Z. Inheritance of long staple fiber quality traits ofinbackground using CSILs., 2012, 124: 1415–1428
[8] Yamamoto T, Yonemaru J, Yano M. Towards the understanding of complex traits in rice: substantially orsuperficially?, 2009, 16: 141–154
[9] Eshed Y, Zamir D. An introgression line population of lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL., 1995, 141: 1147–1162
[10] 王立秋, 赵永锋, 薛亚东, 张祖新, 郑用琏, 陈景堂. 玉米衔接式片段导入系群体的构建和评价. 作物学报, 2007, 33: 663–668 Wang L Q, Zhao Y F, Xue Y D, Zhang Z X, Zheng Y L, Chen J T. Development and evaluation of two link-up single segment introgression lines (SSILs) in., 2007, 33: 663–668 (in Chinese with English abstract)
[11] 高晓清, 谢学文, 许美容, 王磊, 石英尧, 高用明, 朱苓华, 周永力, 黎志康. 水稻抗纹枯病导入系的构建及抗病位点的初步定位. 作物学报, 2011, 37: 1559–1568 Gao X Q, Xie X W, Xu M R, Wang L, Shi Y Y, Gao Y M, Zhu L H, Zhou Y L, Li Z K. Development of introgression lines and identification of QTLs for resistance to sheath blight., 2011, 37: 1559–1568 (in Chinese with English abstract)
[12] Bian J M, He H H, Shi H, Zhu G Q, Li C J, Zhu C G, Peng X S, Yu Q Y, Fu J R, He X F, Chen X R, Hu L F, Ou-Yang L J. Quantitative trait loci mapping for flag leaf traits in rice using a chromosome segment substitution line population., 2014, 133: 203–209
[13] He Q Y, Yang H Y, Xiang S H, Wang W B, Xing G N, Zhao T J, Gai J Y. QTL mapping for the number of branches and pods using wild chromosome segment substitution lines in soybean [(L.) Merr.]., 2014, 12: S172–S177
[14] Korff M V, Wang H, Léon J, Pillen, K. Development of candidate introgression lines using an exotic barley accession (ssp.) as donor., 2004, 109: 1736–1745
[15] Howell P M, Lydiate D J, Marshall D F. Towards developing intervarietal substitution lines innapus using marker- assisted selection., 1996, 39: 348–358
[16] Wang P, Ding Y Z, Lu Q X. Development ofchromosome segment substitution lines in the genetic standard line TM-1 of., 2008, 53: 1512–1517
[17] 朱亚娟, 王鹏, 郭旺珍, 张天真. 利用海岛棉染色体片段导入系定位衣分和籽指QTL. 作物学报, 2010, 36: 1318–1323 Zhu Y J, Wang P, Guo W Z, Zhang T Z. Mapping QTLs for lint percentage and seed index usingchromosome segment introgression lines., 2010, 36: 1318–1323 (in Chinese with English abstract)
[18] 王鹏, 张天真. 利用棉花海陆种间染色体片段导入系剖析光合色素含量的遗传基础. 作物学报, 2012, 38: 947–953 Wang P, Zhang T Z. Genetic dissection of photosynthetic pigment content in cotton interspecific chromosome segment introgression lines., 2012, 38: 947–953 (in Chinese with English abstract)
[19] 付央, 苑冬冬, 胡文静, 蔡彩平, 郭旺珍. 陆地棉背景下海岛棉第18染色体片段置换系的培育及相关农艺性状QTL定位. 作物学报, 2013, 39: 21–28 Fu Y, Yuan D D, Hu W J, Cai C P, Guo W Z. Development ofchromosome 18 segment substitution lines in the genetic standard line TM-1 ofand mapping of QTLs related to agronomic traits., 2013, 39: 21–28 (in Chinese with English abstract)
[20] 黎波涛, 石玉真, 龚举武, 李俊文, 刘爱英, 王涛, 商海红, 巩万奎, 陈婷婷, 葛群, 张金凤, 王永波, 胡玉枢, 袁有禄. 多环境下陆地棉染色体片段代换系及F1皮棉产量与纤维品质的表型分析. 棉花学报, 2016, 28: 75–80 Li B T, Shi Y Z, Gong J W, Li J W, Liu A Y, Wang T, Shang H H, Gong W K, Chen T T, Ge Q, Zhang J F, Wang Y B, Hu Y S, Yuan Y L. Phenotypic analysis of lint yield and fiber quality traits of cotton chromosome segment substitution lines and F1hybrids in multiple environments., 2016, 28: 75–80 (in Chinese with English abstract)
[21] Si Z F, Chen H, Zhu X F, Cao Z B, Zhang T Z. Genetic dissection of lint yield and fiber quality traits ofinbackground., 2017, 37: 9
[22] 肖松华, 刘剑光, 赵君, 吴巧娟, 俞敬忠, 喻德跃. 棉花远缘杂交创制抗黄萎病新种质. 棉花学报, 2015, 27: 524–533 Xiao S H, Liu J G, Zhao J, Wu Q J, Yu J Z, Yu D Y. Creation of a new resistant germplasm towilt by distant hybridization in Upland cotton., 2015, 27: 524–533 (in Chinese with English abstract)
[23] 汪保华, 王为, 庄智敏, 朱新宇. 3个黄褐棉近等基因系的选择及评价. 中国农学通报, 2011, 27: 45–49 Wang B H, Wang W, Zhuang Z M, Zhu X Y. Selection and evaluation of threenear-isogenic lines., 2011, 27: 45–49 (in Chinese with English abstract)
[24] 刘宏伟, 李南南, 苗玉焕, 柳仕明, 聂以春, 朱龙付, 张献龙. 利用FBP:iaaM改良华杂棉H318产量与纤维品质研究. 石河子大学学报(自然科学版), 2016, 34: 134–140 Liu H W, Li N N, Miao Y H, Liu S M, Nie Y C, Zhu L F, Zhang X L. Study on yield and fiber quality improvement of “Huazamian H318” with FBP:iaaM.(Nat Sci), 2016, 34: 134–140 (in Chinese with English abstract)
[25] Kashif I M. 陆地棉×黄褐棉种间全基因组SSR高密度遗传图谱的构建. 中国农业科学院博士学位论文, 河南安阳, 2014 Kashif I M. Genome-Wide SSR High Density Genetic Map Construction from an Interspecific Cross of×. PhD Dissertation of Chinese Academy of Agricultural Sciences, Anyang, China, 2014 (in Chinese with English abstract)
[26] Sun X W, Liu D Y, Zhang X F, Li W B, Liu H, Hong W G, Jiang C B, Guan N, Ma C X, Zeng H P, Xu, C H, Song, J, Huang L, Wang C M, Shi J J, Wang R, Zheng X H, Lu C Y, Wang X W, Zheng H K. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing., 2013, 8: e58700
[27] Shen C, Jin X, Zhu D, Lin Z X. Uncovering SNP and Indel variations of tetraploid cottons by SLAF-seq., 2017, 18: 247
[28] Wang H T, Huang C, Guo H L, Li X M, Zhao W X, Dai B S, Yan Z H, Lin Z X. QTL mapping for fiber and yield traits in upland cotton under multiple environments., 2015, 10: e0130742
[29] Merk H L,Yarnes S C, Van Deynze A, Tong N K, Menda N, Mueller L A, Mutschler M A, Loewen S A, Myers J R, Francis D M. Trait diversity and potential for selection indices based on variation among regionally adapted processing tomato germplasm., 2012, 137: 427–437
[30] Wang B H, Liu L M, Zhang D, Zhuang Z M, Guo H, Qiao X, Wei L J, Rong J K, May O L, Paterson A H, Chee P W. A genetic map betweenand the Brazilian Endemicand its application to QTL mapping.:, 2016, 6: 1673–1685
[31] Zhang T Z, Hu Y, Jiang W K, Fang L, Guan X Y, Chen J D, Zhang J B, Saski C A, Scheffler B E, Stelly D M, Hulse-Kemp A M, Wan Q, Liu B L, Liu C X, Wang S, Pan M Q, Wang Y K, Wang D W, Ye W X, Chang L J, Zhang W P, Song Q X, Kirkbride R C, Chen X Y, Dennis E, Llewellyn D J, Peterson D G, Thaxton P, Jones D C, Wang Q, Xu X Y, Zhang H, Wu H T, Zhou L, Mei G F, Chen S Q, Tian Y, Xiang D, Li X H, Ding J, Zuo Q Y, Tao L N, Liu Y C, Li J, Lin Y Y, Hui Y, Cao Z S, Cai C P, Zhu X F, Jiang Z, Zhou B L, Guo W Z, Li R Q, Chen Z J. Sequencing of allotetraploid cotton (L. acc. TM-1) provides a resource for fiber improvement., 2015, 33: 531–537
[32] Wang J K, Wan X Y, Crossa J, Crouch J, Weng J F, Zhai H Q, Wan J M. QTL mapping of grain length in rice (L.) using chromosome segment substitution lines., 2006, 88: 93–104
[33] Wang J K, Wan X Y, Li H H, Pfeiffer W H, Crouch J, Wan J M. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach., 2007, 115: 87–100
[34] McCouch S R, Cho Y G, Yano P E, Blinstrub M, Morishima H, Kinoshita T. Report on QTL nomenclature., 1997, 14: 11–13
[35] Wang S, Chen J D, Zhang W P, Hu Y, Chang L J, Fang L, Wang Q, Lv F, Wu H T, Si Z F, Chen S Q, Cai C P, Zhu X F, Zhou B L, Guo W Z, Zhang T Z. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes., 2015, 16: 108
[36] 王鹏. 陆地棉TM-1背景的海岛棉染色体片段导入系的培育鉴定和纤维强度QTL精细定位. 南京农业大学博士学位论文, 江苏南京, 2009 Wang P. Development and Evaluation ofChromosome Segment Substitution Lines Cotton in Genetic Standard Line, TM-1 ofand Fine Mapping QTL for Fiber. PhD Dissertation of Nanjing Agricultural University, Nanjing, China, 2009 (in Chinese with English abstract)
[37] Rong J K, Feltus, F A, Waghmare V N, Pierce G J, Chee P W, Draye X, Saranga Y, Wright R J, Wilkins T A, May O L, Smith C W, Gannaway J R, Wendel J F, Paterson A H. Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development., 2007, 176: 2577–2588
[38] Paterson A H, Saranga Y, Menz M, Jiang C X, Wright R J. QTL analysis of genotype × environment interaction affecting cotton fiber quality., 2003, 6: 384–396
[39] Zhang S W, Feng L C, Xing L T, Yang B, Gao X, Zhu X F, Zhang T Z, Zhou B L. New QTLs for lint percentage and boll weight mined in introgression lines from two feral landraces intoacc TM-1., 2016, 135: 90–101
[40] Cao Z B, Zhu X F, Chen H, Zhang T Z. Fine mapping of clustered quantitative trait loci for fiber quality on chromosome 7 using aintrogressed line., 2015, 35: 1–13
QTL Mapping for Yield and Fiber Quality Traits UsingChromosome Segment Introgression Lines
SHEN Chao1, LI Ding-Guo2, NIE Yi-Chun1, and LIN Zhong-Xu1,*
1National Key Laboratory of Crop Genetic Improvement, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, China;2College of Agronomy, Yangtze University, Jingzhou 434025, China
The genetic basis of upland cotton is narrow, which hinders the progress of genetic improvement of cotton. To effectively broaden the genetic basis of upland cotton, we developed BC5S5chromosome segment substitution lines (CSSLs) population consisting of 71 CSSLs, which was derived from the, the wild cotton (AD4) as the donor, and B0011, one parent of national authorized cotton variety Huazamian H318 with good comprehensive characters of upland cotton line (AD1) as the receptor. A comprehensive analysis was conducted via the SLAF-seq genotyping and phenotyping under multiple environments. This population showed a wide range of variation in yield components and fiber quality, and a total of 48 QTLs were detected including 19 for yield components and 29 for fiber quality. Among the QTLs for nine traits, 32 and 16 were on the At and Dt sub-genomes, respectively. Further analysis revealed that 30 QTLs showed positive additive effects, and 18 QTLs showed negative additive effects. The results of this study lay a foundation for the genetic improvement of upland cotton using the elite alleles of important agronomic traits from.
Upland cotton;; CSILs; Yield; Fiber quality; QTL; Additive effect
10.3724/SP.J.1006.2017.01733
本研究由国家转基因生物新品种培育重大专项(2016ZX08009001)资助。
The study was supported by the National Major Project for Developing New GM Crops (2016ZX08009001).
林忠旭, E-mail: linzhongxu@mail.hzau.edu.cn
2017-04-17; Accepted(接受日期): 2017-09-10; Published online(网络出版日期): 2017-09-28.
http://kns.cnki.net/kcms/detail/11.1809.S.20170928.1842.026.html
猜你喜欢
西南农业学报(2022年5期)2022-06-06
中国糖料(2021年3期)2021-07-13
中国中医基础医学杂志(2020年1期)2020-03-03
心电与循环(2020年1期)2020-02-27
中国医学影像技术(2019年10期)2019-10-24
科学之谜(2019年3期)2019-03-28
热带农业科技(2019年1期)2019-01-14
中国果业信息(2019年11期)2019-01-05
科学之谜(2018年8期)2018-09-29
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
