
慢性牙周炎患者龈下菌斑宏转录组菌种分类以及相关代谢基因表达
2018-01-18 05:54张龙张保荣
海南医学 2017年24期
张龙,张保荣
(1.潍坊医学院口腔医学院,山东潍坊261053;2.航空总医院口腔诊疗中心,北京100002)
牙周炎是口腔常见疾病之一,在普通人群中具有较高的发病率。根据不同流行病学调查显示,在普通人群中发病率为80%~90%[1-2]。而慢性牙周炎因其牙槽骨的严重吸收,造成牙齿动度的增加,已成为人类牙齿缺失的首要原因。因此对于慢性牙周炎病因的关注,一直以来是牙周病学的研究热点。传统意义上牙周炎的发生、发展与多种因素相关,比如菌斑、抽烟、遗传、口腔卫生习惯等[3-5],而牙菌斑作为牙周炎的始动因子,在牙周病的发生、发展过程中,发挥了不可替代的重要作用。自一百多年以来,随着各种新的微生物培养技术和生化技术的进步,人们对牙菌斑在牙周炎发病中的作用,有了不断深入的认识,但是采用的传统细菌培养及动物实验,均无法证明其中的一种或多种细菌为牙周炎的明确致病菌。传统方法对细菌及其代谢产物,以及其对人体代谢的影响均基于体外动物实验与相关的生理生化实验[6-7]。而对处于牙周炎疾病过程的菌群变化及其基因表达及代谢产物的分析,受到实验条件以及技术手段限制成为牙周炎病因研究的难点。因此,对于牙菌斑及其细菌及代谢产物在牙周炎发病过程中的处于疾病状态时变化,是探究牙周炎病因的关键。本实验采用基于二代测序技术的对慢性牙周炎患者龈下菌斑的转录组进行分析,首次将龈下菌斑微环境中的整个转录组作为研究对象,从转录组学的高度对慢性牙周炎的病因进行了探讨,为新一代测序技术在慢性牙周炎病因学的研究应用提供了新的思路。
1 材料与方法
1.1 牙菌斑及炎性组织的采集选取航空总医院口腔诊疗中心2015年3月至2016年12月收集的20例慢性牙周炎患者为观察对象,采集龈下菌斑样本采样部位,对所有患者进行牙周炎的程度进行评估:探诊深度、牙周袋深度、附着丧失程度和探针出血情况[8]。纳入标准:3个月内无牙周病治疗史,无抗生素药物服用或其他抗炎药物服用,无吸烟史,无全身性疾病(糖尿病等),妇女无妊娠,全口牙存留不少于16颗,每个区都有存留牙且至少有一个位点的牙周袋深度大于4 mm。所有患者知情同意。采样前嘱患者漱口,用棉签去掉龈上菌斑干扰。每个牙齿均取6个位点[9]。每个样品均溶于1.5 mL HANK液,立即进行-80℃冷冻。
1.2 主要仪器及试剂RNeasy mini kit RNA提取试剂盒(QIAGEN公司)、Illumina HiSeqTM2000(深圳华大)Agilent 2100(安捷伦科技公司上海)Hanks液(北京诺博莱德)干冰若干、冰盒、高速离心机(Thermo Scientific公司,法国)。
1.3 转录组测序前准备
1.3.1 总RNA的提取采用QIAGEN公司的RNeasyMiniKitRNA提取试剂盒。配置所需的了裂解体系Buffer-RLT 500 μL,5 μL B-球基乙醇。取100 μL预处理样品混匀。样品预处理:4℃下解冻,3 min 12 000 r/min离心。取上清液。对于炎性组织则采用液氮研磨后,取上清液。加1:1 70%乙醇洗脱裂解液。12000 r/min 15 s再加入700 μL的Buffer-RW1洗脱。700 μL Buffer-RWE洗脱乙醇(两遍):600 μL RNA-free水洗脱后静置2 min,室温下12 000 r/min、离心2 min。
1.3.2 提取后的RNA浓度将提取后的RNA送交华大基因公司进行上机前提取RNA质量检测评估。提取RNA总量为0.2032~1.3759 μg,RIN值为2.2~2.9,总体符合RNA建库要求[10]。
1.4 上机测序及建库采用深圳华大基因的Illumina HiSeqTM2000进行测序建库[11]。
1.5 测序后数据处理去除含N的碱基数目总和达到一定比例的短序列(默认10%,设置为10%);去除拼接接头污染(默认接头序列与所得序列有15 bp的重叠(设置为15 bp);去除连续低质量值碱基数达到一定长度的序列(默认是序列中小于Q20的碱基数目小于20%,设置为84,20%):宿主相关RNA的去除,将所得原始数据用BLAST软件比对到NCBI的人基因组Nonredundant Database(NR)蛋白数据库[12]去除人相关RNA基因(一致性为90%的前提条件下,与宿主比对上的序列被认为是宿主序列。具体参数设置为:r:1/l:35/M:4/p:2)。所得总的测序可用数据为569782640~8751961080 bp。符合后期分析所需数据要求[13]。
1.6 序列的组装对所得短序列,采用Trinty组装,所得Trinty组装结果我们称之基因序列。组装得到的基因序列,首先使用Tgicl将其去冗余和进一步拼接,然后再对这些序列进行同源转录本聚类,得到最终的非冗余基因序列(Unigene)。可以看出在不同样品中300 bp长度的Unigene数量最多分布范围为2 018~12 906 bp。不同样品组装得到的Unigene聚类软件做进一步序列拼接,去冗余处理以及同源转录本聚类,得到尽可能长的合并非冗余基因序列(merge-Unigene)。在进行同源转录本聚类以后,merge-Unigene分为两部分,一部分是簇merge-Unigene(clusters),同一个clusters里面有若干条相似度高(大于70%)的merge-Unigene(以CL开头,CL后面接基因家族的编号);其余的是单独merge-Unigene(以Unigene开头),代表单独的Unigene。
1.7 基因功能注释基因功能注释主要基于氨基酸序列比对。将基因的氨基酸序列与各数据库进行比对,得到对应的功能注释信息。由于每一条序列比对结果超过一条,为保证其生物意义,保留一条最优比对结果作为该基因的注释。所有的注释均使用BLAST(version 2.2.21)软件结合各个数据库的特点完成。主要采用数据库有:Nonredundant Database(NR):Kyoto Encyclopedia of Genes and Genomes(KEGG)[14-16];Version:59 Carbohydrate-Active Enzymes Database(CAZy)[17];Cluster of Orthologous Groups of proteins(COG)。
2 结果
2.1 慢性牙周炎患者龈下菌斑细菌种类和数量主要细菌为7种:文氏密螺旋体占12.4%、齿垢螺旋体占3.6%、牙龈二氧化碳噬纤维菌占2.8%、牙髓卟啉单胞菌占2.6%、普雷沃菌占2.6%、梅毒螺旋体占2.1%、中间普氏菌占2.0%,而螺旋体占28.1%。
2.2 基于COG数据库的功能注释对比COG中二十五大类功能,与之有比对功能的序列涵盖了其中24种。其中含量最为丰富的是与细菌翻译相关的过程,有超过5 000条merge-unigene,见图1。
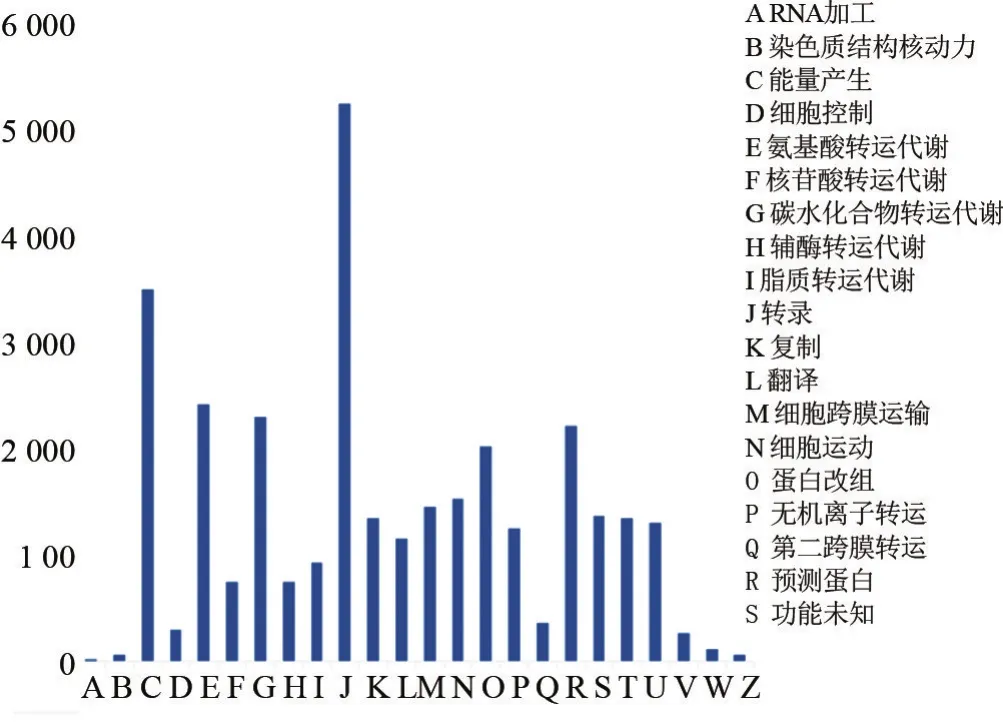
图1 COG数据库merge-unigene功能分析
2.3 基于KEGG数据库的功能注释将merge-Unigene BLAST到KEGG数据库,得到与KEGG数据库中代谢通路相关的merge-Unigene分布情况。在所有样品中主要与之相关的代谢通路为氨基酸的代谢转运、代谢相关。其中数量最多的基因数量集中于精氨酸、脯氨酸代谢通路上,有1 149条merge-Unigene与之相关,见图2。
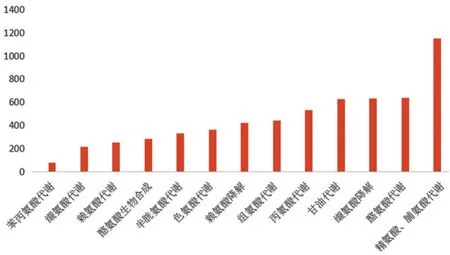
图2 KEGG数据库与氨基酸代谢通路有关的merge-unigene功能
2.4 基于糖类活性酶(carbohydrate-Active Enzymes Database,CAZy)的功能注释merge-Unigene最多的两类碳水化合物酶:碳水化合物结合结构域为43、糖苷水解酶为34、糖苷转移酶为17、碳水化合物酯酶为4,但是与多糖裂解酶相关的merge-unigene数量为0。
3 讨论
慢性牙周炎患者龈下菌斑转录组分析可以看出,在慢性牙周炎患者的龈下菌斑中,传统意义上致病力较弱的螺旋体其基因表达最多,而通常意义上与牙周炎关系密切的牙龈卟啉单胞菌基因表达并不明显。提示了传统方法对于慢性牙周炎的研究有其自身的局限性和不足之处,而通常意义上在研究细菌基因时多关注在DNA层面[18],忽视了RNA水平上基因表达差异的变化。本实验首次抽取龈下菌斑RNA进行了转录组测序,而这个可能为我们解释微量或者低致病性细菌对牙周炎的发生提出新的认识。
本实验的研究结果表明:(1)慢性牙周炎患者龈下菌斑中螺旋体的RNA含量最高,表明螺旋体在牙周炎的疾病过程中基因表达会较其他细菌较多。这与我们通常意义上的牙周炎致病菌认识是不同的[19]。(2)通过转录组分析数据,与不同的蛋白库比对,笔者在整个转录组高度上发现了牙菌斑作为一个整体,在牙周炎的发病过程中与其细菌基本生理活动以及牙菌斑生理代谢过程及通路相关基因的表达情况,并且通过与碳水化合物活性酶相关基因比对,发现龈下菌斑中细菌整体不能利用多糖,但可以通过糖苷水解酶利用单糖及其他双糖等碳水化合物提供强有力的证据[20]。证明单纯的食物残渣进入龈下菌斑,并不能对牙周炎产生致病性。
综上所述,基于高通量的转录组测序分析能为笔者对龈下菌斑致病性的研究提供新思路,并且补充了许多传统实验局限带来的对牙周炎认识的不足之处。提示在牙周炎的发生、发展过程中,螺旋体的基因高表达为牙周炎的发生发展起了推动作用。
[1] Holtfreter B,Kocher T,Hoffmann T,et al.Standards for reporting chronic periodontitis prevalence and severity in epidemiologic studies Proposed standardsfrom the Joint EU/USA Periodontal Epidemiology Working Group[J].J Cin Periodontol,2015,42(5):407-412.
[2] Holtfreter B,Kocher T,Hoffmann T,et al.Prevalence of periodontal disease and treatment demands based on a German dental survey(DMSⅣ)[J].J Cin Periodontol,2010,37(3):211-219
[3] Jenkinson HF,Lamont RJ.Oral microbial communities in sickness and in health[J].Trends Microbiol,2005,13(12):589-595
[4] Moon JH,Lee JH,Lee JY.Subgingival microbiome in smokers and non-smokers in Korean chronic periodontitis patients[J].Mol Oral Microbiol,2014,30(3):227-241
[5] 曹采方.牙周病学[M].北京:人民卫生出版社,2003:127-128.
[6] Wade WG.Has the use of molecular methods for the characterization of the human oral microbiome changed our understanding of the role of bacteria in the pathogenesis of periodontal disease[J].J Clin Periodontol,2011,38(Suppl 11):7-16.
[7] Pihlstrom BL,Michalowicz BS,Johnson NW.Periodontal diseases[J].Lancet,2005,366(9499):1809-1820.
[8] 王丽,吴亚菲,杨禾,等.不同采样方法对龈下牙周致病菌检出的研究[J].牙体牙髓病杂志,2009,19(1):234-238
[9] Maher CA,Kumar-Sinha C,Cao X,et al.Transcriptome sequencing to detect gene fusions[J].Nature,458(7234):97-101.
[10] Logan G,Freimanis GL,King DJ,et al.A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the Illumina MiSeq[J].BMC Genomics,2014,15:828.
[11] Ullmann LS,de Camargo Tozato C,Malossi CD,et al.Comparative Clinical Sample Preparation of DNA and RNA viral nucleic acids for a commercial Deep Sequencing System(Illumina MiSeq<sup>®</sup>)[J].J Virol Methods,2015,220:60-63.
[12] Altschul SF,Gish W,Miller W,et al.Basic local alignment search tool[J].J Mol Biol,1990,215(3):403-410.
[13] Li R,Yu C,Li Y,et al.SOAP2:an improved ultrafast tool for short read alignment[J].Bioinformatics,2009,25(15):1966-1967.
[14] Kanehisa M,Goto S,Kawashima S,et al.The KEGG resource for deciphering the genome[J].Nucleic Acids Res,32(Database issue):D277-D280.
[15] Kanehisa M.A database for post-genome analysis[J].Trends Genet,1997,13(9):375-376.
[16] Kanehisa M,Goto S,Hattori M,et al.From genomics to chemical genomics:new developments in KEGG[J]Nucleic Acids Res,2006,34(Database issue):D354-D357.
[17] Cantarel BL,Coutinho PM,Rancurel C,et al.The Carbohydrate-Active EnZymes database(CAZy):an expert resource for Glycogenomics[J].NucleicAcids Res 2009,37(Database issue):D233-D238.
[18] Wang J,Qi J,Zhao H,et al.Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease[J].Sci Rep,2013,3:1843
[19] Costello EK,Stagaman K,Dethlefsen L,et al.The application of ecological theory toward an understanding of the human microbiome[J].Science,2012,336(6086):1255-1262.
[20] Jacobson RL,Schlein Y,Eisenberger CL.The biological function of sandfly and Leishmania glycosidases[J].Med Microbiol Immunol,2001,190(1-2):51-55
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
现代畜牧科技(2021年4期)2021-07-21
健康之家(2019年9期)2019-12-14
中国动物传染病学报(2019年3期)2019-07-06
意林·少年版(2019年11期)2019-06-30
问健康画报(2017年2期)2017-07-01
中外医学研究(2017年6期)2017-03-30
中华老年口腔医学杂志(2016年5期)2016-03-01
西南医科大学学报(2015年1期)2015-08-22
现代检验医学杂志(2015年1期)2015-02-06
