
催化剂活性位本质和构效关系的模型催化研究
2018-01-13 06:05陈明树
物理化学学报 2017年12期
陈明树
催化剂活性位本质和构效关系的模型催化研究
陈明树*
(厦门大学化学化工学院,固体表面物理化学国家重点实验室,福建 厦门 361005)
明确催化剂的活性位本质和构建多相催化的结构和反应性能之间的准确关系是催化基础研究的重点,表面科学研究基于丰富的表征测试手段能够较好地在分子原子水平测定表面结构以明确催化剂活性位本质,并通过高压原位反应池测定相关催化反应性能,获得较可靠的催化剂构效关系。本文简要总结了近年来本人参与的几个模型催化研究例子,包括贵金属表面上CO和烷烃催化氧化的活性表面、纳米Au膜的制备和CO氧化的催化活性位、VO/Pt(111)上丙烷氧化的协同作用、AuPd合金上醋酸乙烯酯合成Au的助催化作用、模型氧化物上纳米Pt的庚烷脱氢环化制甲苯的粒径关系等,以及相关模型催化研究技术的进展。
模型催化;构效关系;CO氧化;烷烃临氧转化;原位谱学
1 引言
明确催化剂活性位本质和构建准确的催化剂构效关系是多相催化研究的瓶颈难题,这是由于多相催化剂总是同时存在多种活性中心位,其活性、选择性可能有很大差别,催化剂性能评价获取的仅是平均结果。如何准确区分不同催化剂活性中心位的相对活性和选择性,需要结合基于表面结构规整、活性中心位均一的模型催化研究。上世纪六十年代以来,随着真空技术和各种谱学表征手段的发展,模型催化研究借助多功能的表面科学技术在分子原子水平获得很多的表面反应信息,如气体小分子在金属表面发生的化学吸附过程,吸附物之间的相互作用以及可能发生在表面的化学反应1–3,但这些信息大多在接近超高真空(ultrahigh vacuum,UHV)下获得,与真实的催化反应条件存在所谓的“压力鸿沟”。通过对真
空腔体设计改进,Goodman4、Somorjai5等在八十年代后期设计出结合高压反应池与超高真空腔的实验装置,UHV中制备的模型表面可以在UHV中传送到高压反应池中进行接近真实催化反应条件下的催化性能考察和原位谱学表征,克服了“压力鸿沟”,见图1a。
基于单晶表面的研究有助于从分子原子尺度上获得催化剂的构效关系、表面物种等信息4–12。如Ertl等8–10对铁基催化剂上N2吸附活化和与氢作用的详细研究,Somorjai等6,11考察了不同铁单晶面的氨合成相对活性为Fe(111)/Fe(100)/Fe(110) = 418/25/1。Goodman等7,12阐述Ni(111)和Ni(100)表面CO加氢反应测出的反应速率以及表观活化能数值与Ni/Al2O3催化剂相近,表明该反应在 Ni-基催化剂上为非表面结构敏感反应。相较于真实催化剂大多具有复杂的表面结构和较大的比表面积,存在所谓“材料鸿沟”。近年来表面科学研究者进一步致力于研究具有二维规整结构的氧化物薄膜(模型氧化物载体),通过对反模型催化剂的制备和表征13–21,以及在模型氧化物载体上蒸镀金属纳米颗粒等22–25,尽量模拟实际催化剂体系中存在的金属-载体界面结构,可以从模型催化研究更深入地了解真实催化剂体系和真实催化反应过程。
常用模型催化剂的表征手段有测定表面组成、价态、结构等的俄歇电子能谱(AES)和X-光电子能谱(XPS),表面结构的低能电子衍射(LEED)和扫描隧道显微镜(STM),最表面原子层组成的低能离子散射谱(LEIS),表面振动性质、吸附物种等的高分辨电子能量损失谱(HREELS)和镜面反射红外吸收光谱表征(IRAS),表面吸附物的程序升温脱附分析(TPD),反应产物分析的在线质谱和色谱等。
模型催化剂可以选择不同催化活性金属的单晶及不同晶面(含低和高指数面),以及少数半导体氧化物表面(如TiO2、ZnO等),直接研究其表面结构、重构、吸附和催化反应性能,建立催化剂构效关系;也可在金属单晶表面通过物理或化学沉积生长具规整结构的氧化物薄膜,这样可以拓展到包括绝缘体的大多氧化物(如SiO2、Al2O3等),研究其直接作为催化剂活性表面或催化剂载体的性质,同时也可以作为反模型催化剂研究氧化物与金属的相互作用;在模型氧化物(氧化物单晶、氧化物薄膜)表面通过物理或化学沉积制备催化剂活性金属纳米粒子,这种平板型氧化物表面可以很好模拟实际负载催化剂的载体,研究决定催化剂性能的重要参数,如金属与氧化物作用、界面效应、尺寸影响等;金属合金单晶表面、金属单晶表面沉积第二组份,考察合金效应、添加剂的助催作用等26。
催化反应性能评价:UHV中制备并表征后的模型催化剂在真空状态转移至原位反应池进行动力学测试并同时应用IRAS跟踪反应过程中的表面物种,进行一定时间反应后,取一定量的反应混合气进入色谱检测分析。但模型催化剂表面(通常< 1 cm2)的活性中心位极少,比常规多相催化剂性能评价的活性中心位数量低3–6个数量级,催化反应生成产物的量也往往很少,准确定量分析是难点,导致模型催化研究往往局限于催化活性较高的催化剂体系及相关反应、或者仅检测部分产物的生成。目前一些模型催化产物分析可以采用在线质谱跟踪、有些可以跟踪压力变化计算出,较好的定量分析多采用色谱法,其采样方法有:i) 直接色谱取样进样,其进样量通常少于1%的反应池总气体量(灵敏度低);ii) 液氮冷阱捕捉法能使大部分的高沸点组份进入色谱系统进行分析,但对于低沸点物质如H2, N2, CO, O2, CH4等则难以捕捉进样分析。针对这一难点,我们设计了可广泛应用于模型催化反应产物分析的取样进样装置27,如图1(b)所示,该装置主要由一可压缩气袋和可抽真空的不锈钢桶组成,并与反应池和色谱采样六通阀连接,可使反应池中接近80%的气体收集至气袋中,然后压入六通阀采样阀进样。通过真空吸入和加压压缩,可以使反应池总气体量的60%–80%进到高灵敏度气相色谱分析,从而克服液氮冷静捕捉法不能分析低沸点物质的难题,较直接进样法灵敏度提高近两数量级。使模型催化研究拓展至产物生成TOF很低的反应(10−5–10−3molecules per site per second),大大拓展了模型催化研究探针反应的范围。

2 贵金属表面CO催化氧化的活性表面
CO是大多工业环境和室内环境空气中的主要污染物之一,在很多催化反应过程中因反应物带入或反应生成中间物能引起催化反应性能的下降,如痕量CO的存在会引起氢燃料电池电极的中毒,故CO 的选择性消除具有重要意义16,18–25。物理消除法如吸附因效率低、设备庞大,且后处理复杂,限制其广泛应用。化学消除法主要包括催化加氢法和催化氧化法,催化加氢法受化学平衡转化率限制,对低浓度CO的消除效果不佳;催化氧化法在较低温度下使CO氧化生成CO2,是目前尾气处理的最常用方法,同时CO催化氧化也是典型的催化反应,受到广泛的关注。
CO氧化主要的催化剂分为:非贵金属催化剂、分子筛催化剂、合金催化剂、贵金属催化剂。目前主要使用催化剂主要包括非贵金属催化剂如商品化的Hopcalite催化剂(以Cu、Mn的氧化物为活性组分)和以Pt、Pd为活性组分的贵金属催化剂。非贵金属催化剂普遍存在对水蒸气比较敏感,潮湿环境下易失活,室温条件活性较差等缺点。而贵金属催化剂具有催化活性高,稳定性好,寿命长等优点。

图1 (a) 结合高压反应池与超高真空腔的实验装置(Goodman)4;(b)高效模型催化反应进样系统:(1)抽真空待用,(2)负压吸入反应池中气体,(3)压入气相色谱六通采样进样管;含O2, CO, CH4, CO2, C2H4, C2H6, C3H8, C3H6和iso-C4H10标准气体分析谱图,和Pt上丙烷催化氧化的产物分析谱图27
Pt族金属上CO催化氧化是一个典型的催化反应,在近化学计量反应条件下Pt族金属上CO氧化,一般认为遵循Langmuir-Hinshelwood (L-H)机理28–32,稳态反应条件下表面以CO吸附为主,限制了O2的吸附活化,表现为对CO分压接近负一级33–35。在富氧反应条件下,Chen和Goodman等36–39发现在Pt族金属上(包括Pd、Pt、Ph)的CO氧化在O2过量的情况下会出现一个超高活性态,反应速率比计量化学反应条件下高2–3个数量级以上,如图2所示。在超高活性态中,Pd(110)表面用红外吸收光谱检测不到CO吸附峰,其反应速率受CO向表面扩散速率所限制。而基于同步辐射高强度光源发展起来的原位掠角表面XRD、X-射线吸收谱和近常压XPS等的表征,多认为富氧条件下Pt-族金属上CO氧化的活性表面是氧化物表面40–46,当然也有少数学者认为氧化物没有出现47,48。
基于富氧反应条件下Pt-族金属上CO催化氧化存在超高活性和低温活性,其活性表面是金属态表面或氧化物表面是个长期争议的问题,运用原位表征手段研究Pt族金属在富氧条件下活性表面十分重要。陈等应用实验室自行搭建的宽波段镜面反射红外吸收光谱(如图3)37,测量低波数谱带范围(4000–450 cm−1),可以测到大多数金属氧化物的红外特征振动谱带,可以在真实的CO催化氧化条件下对表面变化进行直接观测。研究发现预氧化的Pd表面氧化物在CO中450 K就能被还原,即使在CO/O2混合气中(27 Torr CO和18 Torr18O2)预氧化的Pd(100)表面在450 K时也被还原。表明在CO氧化条件下预氧化的Pt-族金属表面会被还原为金属态表面,也即在CO/O2比值接近化学计量比2/1的条件下,无论是以金属态还是氧化态起始的表面,最终反应的活性表面都是金属表面。

图2 Pd(110)上在O2过量时CO的催化氧化,(a)反应总压和O2/CO分压比值随反应进行的变化, (b) CO和O2分压、CO2生成速率随反应进行的变化36
进而进行釜式反应器研究Pd单晶上CO的氧化37,CO的反应速率由跟踪反应总压变化计算获得,并应用同位素18O2来进一步考察在反应条件下PdO是否形成。反应温度为525 K,O2/CO的初始比值为2,由于O2过量18O2/CO的比值随反应进行而不断增加。可以看出随反应时间的增长到一定的O2/CO比时反应压力突然急剧下降,表明反应速度的突然加快,也即进入高活性态,同时红外吸收光谱中CO2的气相峰强度也急剧增加。用液氮冷井直接连接在反应容器上可进一步净化反应气体的同时可部分除去反应生成的CO2。由于Pd18O的主红外吸收谱带648 cm−1与反应过程中生成的气相18OC16O的弱吸收峰648和644 cm−1重叠,影响表面PdO的检测。为评价反应过程中是否有PdO生成,把表面Pd18O谱图和气相18OC16O谱图叠加在一起,如图4所示,在648 cm-1附近的肩峰(也就是PdO+CO2)有显著增加,故可以通过计算谱峰648和644 cm-1强度的平均值(相当于surface+CO2)和气相CO2左边肩峰677 cm−1谱峰强度(相当于CO2)的比值,与纯气相CO2的比值对比就可以衡量CO氧化过程中Pd表面氧化物是否生成。所得比值与反应时间的关系图如图4所示,在整个反应过程中,包括低活性区域和高活性区域,这个比率都基本上是恒定在0.97,明显低于PdO和CO2的加和谱的比值1.11。因此使用同位素18O2在CO氧化的原位IRAS实验表明PdO在超高活性区域并没有形成。事实上,在富氧条件下经过超高活性区域的样品在冷却至室温,抽至真空后得到的红外谱图与PdO谱峰相比,也没有明显的氧化物生成。
进而考察了更加接近于真实催化剂体系的多晶Pd片上CO氧化,反应温度降低到500 K,尽可能使反应生成CO2的速率接近于液氮移除CO2的速率,使反应池中CO2的分压降低,以更为准确评价CO氧化反应的活性表面。图5是在实际CO氧化过程中750–550 cm−1区域的红外谱图。PdO的红外振动谱峰和CO2的振动谱峰比较接近,即使使用同位素18O2,依然很难从气相CO2的振动模式中区分出PdO的振动模式。用压力刚开始骤降的谱图为背景扣超高活性区域后的谱图,得到如图5,谱线20和21的气相CO2峰强度基本可以消除。比较谱线21和多晶Pd片在500 K 10 Torr O2氧化10 min得到的PdO/Pd谱线以及Pd(100)在700 K 10 Torr O2氧化5 min得到的PdO谱线,在富氧条件下CO氧化500 K并没有检测到表面氧化物的生成。在超高活性态时CO2会连续增加,CO气相则连续减少以及在1970 cm−1处CO吸附峰的消失。在超高活性态CO吸附峰的消失和在Pd(110)上观测到的结果一致。在超高活性区域后观测不到CO气相,这也说明在超高活性区域CO耗完,且反应速率受CO的扩散速率所影响,这也被Chen等前期的动力学研究所证实36,49,50。

图4 (a)固定反应容器中反应总压、反应温度和反应时间的关系。起始反应条件为:45 Torr气体 (CO/O2∼1/2)。反应器是和一个可以持续补给气体的容器相连,补给速度远低于反应速率,同时连接一个液氮冷井净化反应气并可部分移除反应生成的CO2,(b) PdO表面, 气相18OC16O, 以及 PdO + 18OC16O IR谱图,(c) 648 + 644 cm−1谱峰强度与677 cm−1谱峰强度比率于反应时间关系37

图5 多晶Pd上CO氧化反的红外光谱图。(a)高波数区观测气相CO2、CO和表面吸附CO,(b)低波数区观测气相CO2的弯曲振动和催化剂表面变化37
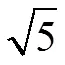
基于模型催化研究结合IRAS系统并结合同位素技术(18O2)研究,充分证实了Pt-族金属表面在实际CO氧化过程中的活性表面不是氧化物表面而是CO(低活性区)或化学吸附氧覆盖(高活性区)的表面(如图6所示)。而基于X-射线相关的原位实验导出不同结论很可能与其原位池中检测表面所处环境、而且与X-射线的光子能量足够使O2分子解离成原子氧而使Pt-族金属易于被氧化有关,如Gottfried的实验确实发现在近常压XPS实验中Au纳米粒子也可被O2氧化54。
3 Au催化剂上CO氧化
1987年Haruta等55–58发现负载在合适的氧化物载体上的Au小纳米颗粒(<5 nm),在低于室温条件下就具有催化CO氧化的活性。随后Au作为低温CO氧化催化剂备受关注,研究发现Au纳米催化剂的性能对反应条件比较敏感,且依赖于催化剂的制备方法、载体类型、Au粒径及量子尺寸效应、预处理条件和存放条件、残存的杂质(特别是氯离子含量)。Haruta等55–58认为Au0是反应的活性位,金属-载体界面对O2活化起到重要作用,3 nm的Au有较好的CO催化氧化活性,活性与粒径存在火山型曲线。Goodman等22,59将Au沉积到TiO2(110),发现具有3层以上(含3层)的Au纳米簇表现为金属特性,单个原子厚度的Au簇具有相当大的带隙表现为绝缘体性质,2个原子厚度的Au簇则表现为由金属特性向非金属特性转变的过渡态并具最高的室温CO催化氧化活性,纳米Au粒子的活性与尺度呈火山型曲线。近年张涛等发现很低负载量下Au以单原子分散在FeO等表面也表现出较好的CO氧化活性,如0.05%Au/Co3O4在室温时可获得TOF ≈ 260。
Chen等25,61–66成功合成并表征了具结构规整性的模型氧化物载体TiO/Mo(112),并进而在该模型氧化物载体上成功合成具单/双原子层厚度的结构规整的金纳米膜(图7)。CO催化氧化活性测试表明,所合成的双原子层金纳米膜具有很高的催化活性,其催化活性较常规氧化物负载的金纳米颗粒(Au/TiO2)高几十到上百倍。规整结构的双原子层金纳米膜具有显著高于单原子层膜的催化反应活性,充分表明具特殊催化性能的金纳米催化剂的活性位和活性相是与氧化物载体接触的第一和第二层金原子的结合。随后Kung等的研究也发现纳米Au周边原子起关键作用67。上述模型催化研究居于确凿的实验证据不仅成功地揭示纳米金催化剂的活性位本质,而且展示出金纳米膜催化剂具有比金纳米颗粒更高的催化活性。
基于模型催化研究结果,陈等尝试在常规SiO2载体上用化学嫁接方法生长单层或亚单层TiO膜,并进而负载金,发现比常规SiO2、TiO2负载的金催化剂其催化性能大为提高68,69。
4 CH4部分氧化

图6 Pt-金属上不同表面相CO氧化的相对活性比较39
CH4部分氧化的催化剂活性相的判定以及催化反应机理究竟是遵循直接氧化机理还是燃烧重整机理等问题目前都仍存在争议70–75。在Pt族金属Pd、Rh和Pt基催化剂上CO或H2的氧化反应较甲烷容易36–39,直接氧化机理生成的CO和H2很有可能在催化剂床层前部有O2区域进一步被氧化生成CO2和H2O;而在催化剂床层后部无O2区域,CH4与CO2和H2O发生重整反应生成CO和H2。除催化剂床层这样的浓度分布外,CH4氧化反应是一个强放热反应,而重整反应是吸热反应,导致催化剂床层存在温度梯度,动力学测试难以获得直接关联信息。

图7 Mo(112)-(8 × 2)-TiOx负载单、双原子层Au膜的CO催化活性和STM图25,模型载体和高比表面积载体负载的金纳米颗粒的一氧化碳催化氧化活性
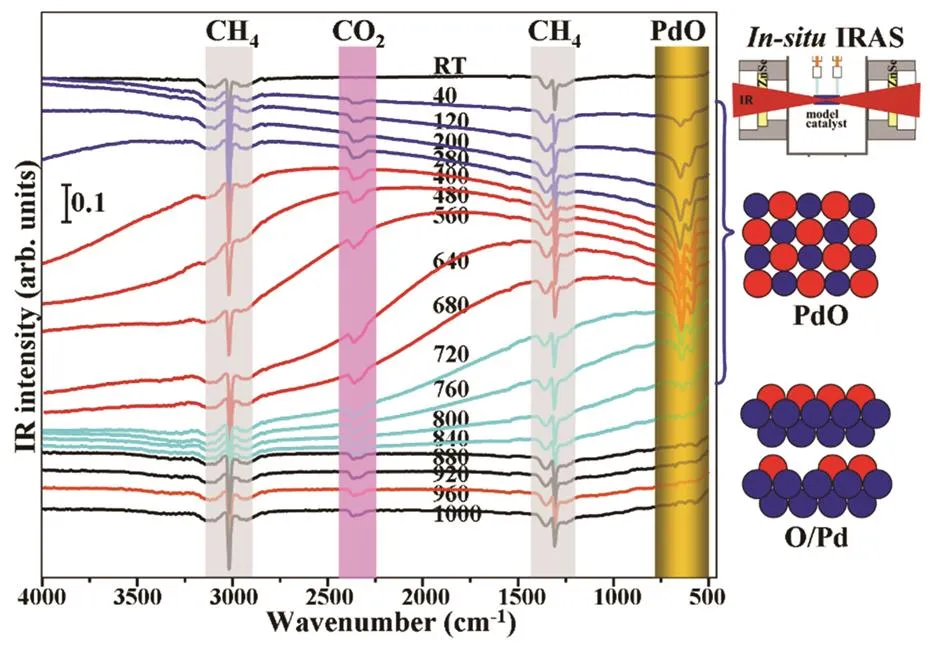
图8 多晶Pd金属表面上643 K CH4氧化反应随时间变化的原位红外反射吸收光谱图及谱峰归属示意图 (起始反应物总压10 Torr,初始CH4/O2 = 9/1)75

图9 釜式反应器中甲烷在金属Pd表面氧化的反应压力、CH4和O2分压、反应速率随反应时间变化(643 K, 9 Torr CH4, 1 Torr O2)。插图是从原位红外光谱得出的活性表面示意图75
陈等75采用Pd和Pt多晶或单晶金属表面作为模型催化剂可以克服催化剂床层的浓度和温度梯度问题,同时添加液氮 (78 K) 冷阱(CO2和H2O的饱和蒸气压低于10−6Torr,CH4,CO,O2的约为10.2 Torr、160 Torr和360 Torr,H2为气态)移去CO2和H2O以尽量减少重整反应的干扰,实验所用反应温度较通常POM反应所用的温度低,也可以排除重整反应。并结合宽波段原位红外光谱实时跟踪Pd和Pt的表面变化,在真实催化反应条件下观察到了多晶Pd金属表面的氧化/还原变化并且将其与催化活性相关联。通过高灵敏度在线色谱的检测确定了所考察实验条件下主要产物为CO2和H2O,有
CH4(g) + 2O2(g) = CO2(s, 78 K) + 2H2O(s, 78 K)
故在釜式反应器中通过测定压力变化可以推算出反应速率。
在多晶Pd金属表面上,643 K贫氧条件(初始CH4/O2= 9/1)下CH4氧化反应过程中,原位红外光谱(图8)观测到600和645 cm−1处可归属于PdO的红外振动吸收峰,其反应TOF较高(图9)。随着反应的进行O2分压迅速下降,PdO逐渐被还原,TOF也随之降低。继续反应,TOF反而有所增加,并很快达到一个峰值,也即超高活性状态,其对应的CH4/O2分压比值随反应温度升高而降低。
比较多晶Pt金属表面上CH4氧化反应总压与反应时间变化的关系(图10a),与CO氧化的变化很相似(图10b)。富氧条件下多晶Pd金属表面上CO氧化反应初期反应速率(TOF)较低,表面主要由化学吸附的CO所覆盖,对O2的解离吸附较为困难,此时反应速控步骤为CO的脱附为O2提供活化吸附位;随着反应进行,CO分压大幅度下降,吸脱附平衡导致表面CO覆盖度有所降低,O2/CO比值增大,O2在表面活化解离概率增大,反应速率提高;当表面化学吸附的O原子覆盖度增加到一定值时即达到了超高活性状态,TOF骤增并且反应温度在短时间内有所上升。750 K贫氧条件下多晶Pt金属表面上CH4氧化随着反应的进行总压逐步增快,即反应TOF随着O2分压的减小在逐渐增大。在该实验所研究的温度范围和气体压强范围内,原位红外光谱和拉曼光谱表征结果都没有检测到Pt氧化物物种。该现象进一步说明了Pt表面上CH4氧化反应与Pd表面上CO氧化反应一样遵循L-H机理,即化学吸附O与CH物种在催化剂Pt表面竞争吸附,CH4/O2接近化学计量比时Pt表面被化学吸附O所覆盖,此时反应速控步骤为CH4的吸附活化。多晶Pd和Pt金属表面上CH4氧化反应中观察到的超高活性状态对应的TOF主要是取决于反应物O2剩余量以及传质扩散的限制,表明金属态Pd表面CH4氧化反应同样遵循L-H机理,CH4与O2在Pd表面竞争吸附,而金属Pd活化O2的速度较之活化CH4的速度快得多。

图10 釜式反应器中(a)甲烷在金属Pt表面(750 K, 10 Torr CH4, 3 Torr O2) , (b)CO在Pd表面(523 K, 2.3 Torr CO, 2.7 Torr O2(氧化的反应压力、反应速率随反应时间变化。插图是从原位红外光谱得出的活性表面示意图75
Fig.10 The changes of the total pressure and reaction rate as functions of the reaction time for (a) CH4oxidation on Pt, (b) CO oxidation on Pd75.
通过上述模型催化研究探明:i) PdO是甲烷燃烧反应的一种高活性表面与文献报道吻合76,77;ii)金属态的Pd和Pt表面在通常反应条件下主要为化学吸附氧饱和、抑制CH4的活化,活性较低;iii)在很低氧分压条件下金属态的Pd和Pt表面化学吸附氧覆盖度不足盖满单层而暴露部分未覆盖金属表面具有高于PdO表面的活性;iv)裸露的表面Pd和Pt是活化CH4的关键,反应遵循Langmuir-Hinshelwood机理。
5 VOx/Pt(111)模型催化剂上丙烷氧化性能研究
能源与环境是社会可持续发展面临的巨大挑战,丙烷催化氧化燃烧反应对提高丙烷利用率,减少挥发性有机化合物(VOCs)排放具有重要意义。在丙烷催化氧化燃烧催化剂中,贵金属Pt催化剂因其低温燃烧活性受到了广泛研究。许多研究结果表明Pt-基催化剂的性能与载体、助剂、制备方法以及处理条件等因素有关。钒基催化剂是碳氢化合物临氧定向转化的重要工业应用催化剂,其选择性和催化活性的提高对资源优化利用和节能减排具有极为重要的意义78–85。由于钒基催化剂的价态变化和分散不均一,谱学上各钒氧物种特别是V-O-V和V-O-S的鉴别尚不清晰。为此我们开展了VO/Pt(111)模型催化研究。
VO/Pt(111)模型表面的制备采用蒸着-氧化法制得86,87,在1 × 10−7Torr O2气氛下623 K氧化,可以获得层层生长至三个单层以上氧化物膜。HREELS结果(图11)表明0.5 ML以下表面钒氧化物的声子振动主要位于128–129 meV,归属于表面钒氧双键(V=O)的伸缩振动;高于0.5 ML,出现可归属于V–O–V的声子振动的69 meV;当VO覆盖度超过1 ML后,表面开始出现位于62 meV的新振动峰,随着覆盖度的增加,位于62 meV的新振动峰强度逐渐增强,而位于69 meV的声子振动强度逐渐减弱,在2 ML时完全消失,对应于一种双层钒氧化物膜结构;2ML以上又出现对应V=O双键的振动峰。
图12为373、423以及473 K时,Pt(111)表面丙烷反应速率随VO覆盖度变化的曲线图27。由图可以看出,清洁的Pt(111)表面有一定的丙烷催化氧化活性,反应产物为CO2,测试条件下没有检测到甲烷、乙烷、乙烯、丙烯以及CO等其他含氧化合物。表面蒸镀亚单层VO后,373 K时的丙烷转化速率随VO覆盖度的增加而迅速降低,而在423和473 K 时丙烷转化速率随VO覆盖度的增加表现出先增加后降低并在VO覆盖度为约0.3 ML时获得最佳丙烷转化速率。
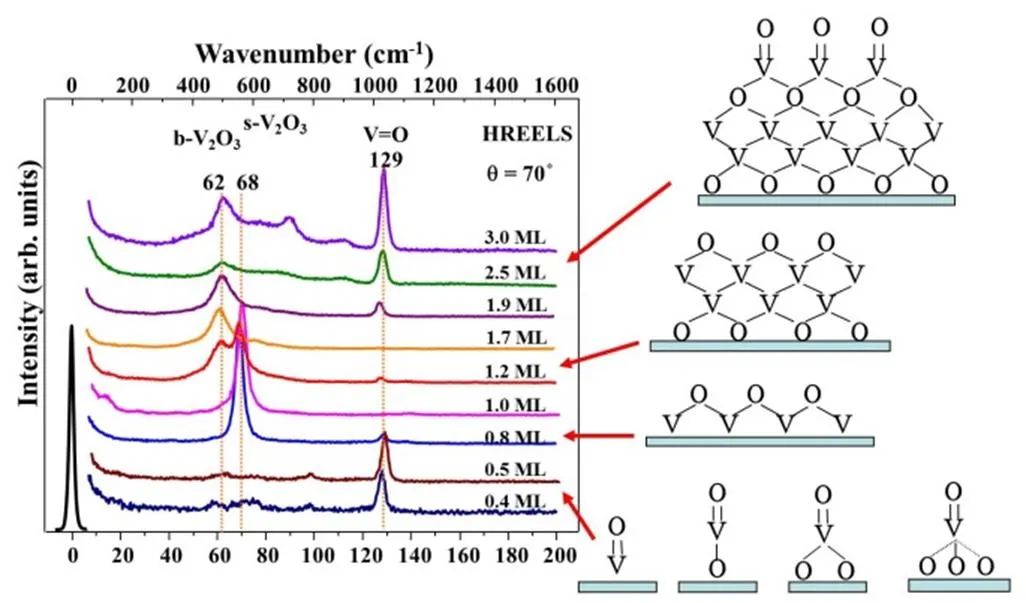
图11 Pt(111)表面不同覆盖度的VOx的HREELS谱图87

图12 不同反应温度下Pt(111)表面丙烷反应速率随VOx覆盖度变化的曲线图27
C3H8:O2= 1:1,total= 4 Torr
应用镜面反射红外吸收光谱(IRAS)比较0.9和3 ML的VO/Pt(111) 模型表面在O2、C3H8以及O2-C3H8混合气中的变化(图13)。0.9 ML的VO/Pt(111) 模型表面暴露在1 Torr氧气中,室温下,观察到位于1038 cm−1可归属于V=O振动的生成,随着暴露时间增长V=O的振动强度不断增强,表明VO在更高O2分压下被进一步氧化生成高价矾氧化物并含V=O,这也与准原位XPS测定结果吻合。该氧化后的0.9 ML的VO/Pt(111) 表面暴露在1 Torr C3H8气氛下,室温即可观察到生成的V=O被C3H8还原。将被丙烷还原后的表面,暴露在含有1 Torr氧气和1 Torr丙烷混合气体中,室温下表面V=O很快就能恢复。表明0.9 ML VO/Pt(111)模型表面具有很强的氧化还原能力。而3 ML的VO/Pt(111) 模型表面同样暴露在1 Torr氧气中后需在较高温度下(373 K)才能观察到位于1038 cm−1的出现,同样氧化后生成的V=O物种也需在较高温度下(403 K)才能被还原。表明0.9 ML VO/Pt(111)表面较3 ML VO/Pt(111)的模型表面具有更活泼的氧化还原性质,可能与其较高的丙烷催化氧化活性有关。
实际上Pt催化剂本身即具有一定的丙烷低温燃烧性能82–85,88,但由于氧的活化吸附比丙烷容易,低温有氧条件下表面几乎被化学吸附氧覆盖,抑制了丙烷的活化,表现出较低的反应速率75。钒氧化物的添加,可能因其存在的酸性位能提高丙烷在表面的吸附强度82,85,但VO位上丙烷的活化比在清洁Pt表面活化能高,只有在比较高的反应温度下才有明显的促进作用。但是值得注意的是,这种促进作用并不随着VO覆盖度的增加线性增加,而是在覆盖度约为0.3 ML时其促进作用达到最大值,当覆盖度超过一个单层后,其反应速率则低于干净的Pt(111)表面,表现出抑制作用,与RAIRS表征结果一致。表明VO-Pt界面间存在协同作用:Pt表面吸附活化氧,与临近VO上吸附丙烷反应。

图13 0.9 ML (上)和3 ML (下) VOx/Pt(111)在不同气氛的原位红外光谱图27:(A,D) 1 Torr O2,(B,E) 1 Torr C3H8, 和(C,F) 1 Torr O2 + 1 Torr C3H8
6 合金催化剂
金属的特性会因为加入别的金属形成合金而改变,它们对化学吸附的强度、催化活性和选择性等效应都会改变,对其稳定性也有影响26,89–93。如炼油工业中Pt-Ir、Pt-Re及PtSn重整催化剂的应用,大大提高了芳构化的活性和选择性,开创了无铅汽油的主要来源,给催化重整技术带来了一个“革命”性的提高89。又如汽车尾气催化燃烧所用的Pt-Rh及Pt-Pd催化剂,为防止空气污染作出了重要贡献。合金催化剂的性能促进主要来自组合成分间的协同效应,包括几何效应和电子效应。合金化不仅能改善催化剂的选择性,也能促进稳定性。例如,轻油重整的Pt-Ir催化剂,较之Pt催化剂稳定性大为提高。其主要原因是Pt-Ir形成合金,避免或减少了表面烧结。Ir有很强的氢解活性,抑制了表面积炭的生成,维持和促进了活性。
Pd-Au合金催化剂上醋酸乙烯酯单体的合成是重要的化工原材料生产途径26,92,93。Au组份基本无活性,Pd是活性催化剂组份,但单用Pd组份其醋酸乙烯酯的生成选择性很差,而将两者结合其活性和选择性均大大提高。该过程虽然已经工业化了,但Au的助催化作用仍不清楚。陈等26,93用对该反应无催化活性的Au(111)和Au(100)单晶表面作为模型催化剂的基底构筑Pd-Au合金表面,将Pd蒸着到Au表面再通过真空退火可以使表面Pd原子完全被Au所孤立,CO-IRAS结果表明CO可以在Pd的桥位或穴位上吸附,当Pd完全被Au孤立后只观测到Pd顶位上的吸附峰。在Pd/Au(111)和Pd/Au(100)上进行醋酸和乙烯氧化制醋酸乙烯酯反应(图14)发现其产物生成速率随Pd覆盖度的降低而增大,在Au(100)面上当Pd覆盖度低于0.1 ML后产物生成速率反而降低,Pd/Au(100)上的活性比Pd/Au(111)的大得多。
该模型催化研究结果揭示醋酸乙烯酯单体合成的Pd-Au合金催化剂的活性表面和活性位的结构分别是fcc(100)晶面和该面上的一对被Au原子隔离的孤立Pd原子,阐明Au的助催化作用机制主要是隔离多Pd原子活性位以形成单原子Pd活性中心(图14),削弱反应中间物或产物CO的吸附,而提高催化剂抗中毒能力,同时消除反应物分子在催化剂表面上深度分解的途径(毗邻Pd原子簇上乙烯的逐步裂解),有效地抑制副产物的形成而提高选择性并进而提高活性。完全被助催剂原子分割的单一活性金属原子作为催化活性位概念的提出对于许多催化选择性反应、助催剂的促进作用等的诠释具有重要的指导意义,从而对多组分催化剂的改进、新型催化剂的研制具有重大的应用价值。
7 Pt/SiO2模型催化剂上庚烷脱氢环化研究
构建多相催化剂的结构和反应性能之间的准确关系是催化研究的重点,金属单晶因其规整的表面结构可以获得较好的构效关系,但很多催化反应对表面结构、尺寸敏感,并且与载体之间存在相互作用,单晶表面与真实催化体系存在所谓的“材料差异”。Goodman等94–96构筑常规催化剂载体TiO2,Al2O3和SiO2等的模型表面,并在其上蒸着金属纳米粒子,以模拟复杂的实际催化剂体系,并以庚烷脱氢环化制甲苯做探针反应。脂肪烃的催化脱氢环化反应是石油化工中的重要反应之一,用于提高汽油的辛烷值97。通过CO-IRAS和程序升温脱附(TPD)测定表面台阶位和平台位数与Pt粒径大小的关系98,并进而详细考察其对庚烷脱氢环化反应性能的影响,建立反应速率与表面平台位、台阶位和纳米颗粒尺寸之间的关系。
在Pt/SiO2/Mo(112)模型催化剂上,Pt纳米粒子尺寸显著影响庚烷脱氢环化反应速率(图15)99,随颗粒度的减小反应速率增大,在1.5 nm时达到最大,颗粒度继续减小反应速率急剧下降。从CO-IRAS、CO-TPD和理论模拟结果Pt纳米粒子的表面同时存在台阶位和平台位,其中台阶位占总表面位的比值随颗粒度减小而增多,但平台位在很小粒子上将不存在。反应速率和Pt粒径变小不是单调增加的趋势,表明脱氢环化反应需要有特定配位环境的活性中心位。这也反映在Pt(110)和Pt(100)两个单晶活性都比Pt纳米颗粒低了近一个数量级,表明拥有C8平台面的Pt(100)和主要为台阶位的Pt(110)单晶面其生成甲苯的这个反应中活性并不高。考虑庚烷的脱氢环化需要断裂C―H键,很多研究表明配位不饱和的台阶位上C―H键解离的活化能低,故纳米粒子越小其台阶位密度越大脱氢活性越高;但环化反应在平台位上比较有利,很小颗粒的平台位减少并趋于零,表面原子簇结构与六个碳原子的环化不配,活性反而低。Gillespie等100也认为庚烷的脱氢环化反应在既有平台位(如C9,C8配位)又有台阶位(如C7,C6配位)的时候活性更好,故表面的活性位必须是在平台-台阶界面的配位不饱和位。
Pt/SiO2/Mo(112)模型催化剂上庚烷脱氢环化研究揭示其结构敏感性问题,建立明确的结构与性能的关系,成功展示了模型催化研究对解决多相催化反应机理的重要性。
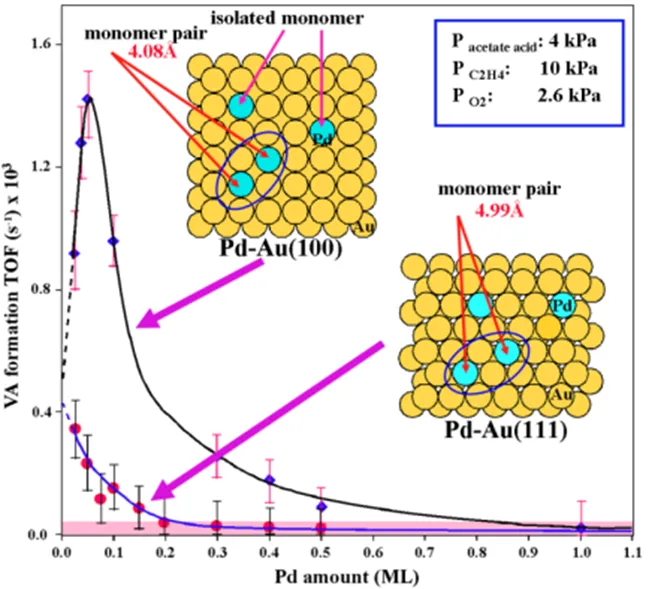
图14 Pd/Au(100)和Pd/Au(111)上醋酸乙烯酯合成速率与Pd覆盖度的关系26
8 结论与展望
上述几个模型催化研究例子展示模型催化研究能够克服常规多相催化研究存在的固有内在难题,即表面的复杂性导致难以准确判定活性中心位和活性反应中间体,成功揭示几个反应的催化剂活性中心位/活性表面和建立较为准确的催化剂构效关系。但目前模型催化的研究大多仅局限在金属的单晶表面,主要是因研制具结构规整性的模型催化剂表面,特别是氧化物载体负载的金属模型表面难度大,需有较大的仪器设备投入和海选模型催化剂表面的制备方法。另一个限制模型催化研究的因素是模型催化剂上反应产物的检测,因为单晶的表面积通常仅~1 cm2,远小于粉末催化剂(~100 m2∙g−1),故模型表面催化反应产物的量很少,同时反应池壁的催化贡献以及反应原料气中可能含有的微量产物通常造成比较高的背景值,导致模型催化剂表面催化反应产物的准确检测仍存在困难。

图15 (a)庚烷脱氢环化制甲苯生成速率与Pt纳米颗粒尺寸的关系,(b) CO TPD (黑点)和硬球模型(红线)对Pt纳米颗粒CUS位的量化99
因此借助现代表面科学手段,通过模型催化剂表面的设计/合成,研制对有关反应催化惰性以及尽量小体积的原位反应池,以建立准确的结构~催化性能关系;研发仅对表层敏感的原位表面组分测试(原位低能离子散射谱)和原子分辨谱学(针尖增强拉曼光谱)技术等,结合多种原位谱学/显微镜技术,并重视应用高时间分辨率的技术和方法,应用以量子化学为主的理论方法,在原子、分子水平上认知催化剂活性位本质和反应机理,比较和关联不同催化剂模型表面上特定化学键活化模式和选择性的控制因素,将会大幅提高催化学科的研究水平和在分子水平上的催化剂设计/研制。
(1) Somorjai, G. A.; Li, Y.; John Wiley & Sons: NJ, USA, 2010.
(2) Ertl, G.; Freund, H.-J.1999,, 32. doi: 10.1063/1.882569.
(3) Freund, H.-J.; van Santen, R. A.; Neurock, M.; Boudart, M.; Mullins, C. B.; Norskov, J. K.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; pp. 911–1051.
(4) (a) Goodman, D. W.. 1994,, 837. doi: 10.1016/0039-6028(94)90701-3 (b) Goodman, D. W.1996,, 13090. doi: 10.1021/jp953755e
(5) Cremer, P. S.; Su, X. C.; Somorjai, G. A.; Shen, Y. R.1998,, 225. doi: 10.1016/S1381-1169(97)00268-9
(6) Spencer, N. D.; Schoonmaker, R. C.; Somorjai, G. A.. 1982,, 129. doi: 10.1016/0021-9517(82)90016-1
(7) (a) Goodman, D. W.; Kelley, R. D.; Madey, T. E.; Yates, J. T., Jr.1980,, 226. doi: 10.1016/0021-9517(80)90075-5 (b) Goodman, D. W.1982,, 522. doi: 10.1116/1.571422
(8) Ertl, G.2008,, 3524. doi: 10.1002/anie.200800480
(9) Grunze, M.; Bozso, F.; Ertl, G.; Weiss, M.1978,, 241. doi: 10.1016/0378-5963(78)90017-X
(10) Bozso, F.; Ertl, G.; Weiss, M.. 1977,, 519. doi: 10.1016/0021-9517(77)90063-X
(11) Strongin, D. R.; Carrazza, J.; Bare, S.R.; Somorjai, G.A.1987,, 213. doi: 10.1016/0021-9517(87)90109-6
(12) Kelley, R. D.; Goodman, D. W.1982,, L743.
(13) Belton, D. N.; Sun, Y. M.; White, J. M.1984,, 1690. doi: 10.1021/j150653a005
(14) Ocal, C.; Ferrer, S.1986,, 6474. doi: 10.1063/1.450743
(15) Bradford, M. C. J.; Vannice, M. A.1997,, 31. doi: 10.1023/A:1019022903491
(16) Martynova, Y.; Shaikhutdinov, S.; Freund, H.-J.. 2013,, 2162. doi: 10.1002/cctc.201300212
(17) Willinger, M. G.; Zhang, W.; Bondarchuk, O.; Shaikhutdinov, S.; Freund, H.-J.; Schlögl, R.2014,, 5998. doi: 10.1002/anie.201400290
(18) Fu, Q.; Li, W. X.; Yao, Y. X.; Liu, H. Y.; Su, H. Y.; Ma, D.; Gu, X. K.; Chen, L. M.; Wang, Z.; Zhang, H.;2010,, 1141. doi: 10.1126/science.1188267
(19) Chen, G. X.; Zhao, Y.; Fu, G.; Duchesne, P. N.; Gu, L.; Zheng, Y. P.; Weng, X. F.; Chen, M. S.; Zhang, P.; Pao, C. W.;.2014,, 495. doi: 10.1126/science.1252553
(20) Yao, Y. X.; Fu, Q., Zhang, Y. Y.; Weng, X. F.; Li, H.; Chen, M. S.; Jin, L.; Dong, A. Y.; Mu, R. T.; Jiang, P.;.2014,, 17023. doi: 10.1073/pnas.1416368111
(21) Zhang, Y. H.; Weng, X. F.; Li, H.; Li, H. B.; Wei, M. M.; Xiao, J. P.; Liu, Z.; Chen, M. S.; Fu, Q.; Bao, X. H.2015,, 3616. doi: 10.1021/acs.nanolett.5b01205
(22) Valden, M.; Pak, S.; Lai, X.; Goodman, D. W.1998,, 7. doi: 10.1023/A:1019028205985.
(23) Chen, M. S.; Goodman, D. W.2008,, 264013. doi: 10.1088/0953-8984/20/26/264013
(24) Schauermann, S.; Nilius, N.; Shaikhutdinov, S.; Freund, H.-J.2013,, 1673. doi: 10.1021/ar300225s
(25) Chen, M. S.; Goodman, D. W.2004,, 252. doi: 10.1126/science.1102420
(26) Chen, M. S.; Kumar, D.; Yi, C. W.; Goodman, D. W.2005,, 291. doi: 10.1126/science.1115800
(27) Zheng, Y. P.; Zhang, L. H.; Wang, S. L.; Tang, Z. Y.; Ding, D.; Chen, M. S.; Wan, H. L.2013,, 9090. doi: 10.1021/la401256z
(28) Langmuir, I.1922,, 621.
(29) Berlowitz, P. J.; Peden, C. H. F.; Goodman, D. W.1988,, 5213. doi: 10.1021/j100329a030
(30) Engel, T.; Ertl, G.;1979,, 1. doi: 10.1016/S0360-0564(08)60133-9
(31) Campbell, C. T.; Ertl, G.; Kuipers, H.; Segner, J.1980,, 5862. doi: 10.1063/1.440029
(32) Kim, S. H.; Mendez, J.; Wintterlin, J.; Ertl, G.2005,, 155414. doi: 10.1103/PhysRevB.72.155414
(33) Stuve, E. M.; Madix, R. J.; Brundle, C. R.1984,, 155. doi: 10.1016/0039-6028(84)90235-8
(34) Ozensoy, E.; Meier, D. C.; Goodman, D. W.2002,, 9367. doi: 10.1021/jp020519c
(35) Kuhn, W. K.; Szanyi, J.; Goodman, D. W.1992,, L611. doi: 10.1016/0039-6028(92)90834-S
(36) Chen, M. S.; Cai, Y.; Yan Z.; Gath, K. K.; Axnanda, S.; Goodman, D. W.2007,, 5326. doi: 10.1016/j.susc.2007.08.019
(37) Chen, M. S.; Wang, X. V.; Zhang, L. H.; Tang, Z. Y.; Wan, H. L.2010,, 18113. doi: 10.1021/la103140w
(38) Chen, M. S.; Zheng, Y. P.; Wan, H. L.2013,, 1299. doi: 10.1007/s11244-013-0140-0
(39) Weng, X. F.; Yuan, X.; Li, H.; Li, X. K.; Chen, M. S.; Wan H. L.2015,, 174. doi: 10.1007/s11426-014-5277-6
(40) Hendriksen, B. L. M.; Bobaru, S. C.; Frenken, J. W. M.2004,, 229. doi: 10.1016/j.susc.2004.01.025
(41) Hendriksen, B. L. M.; Frenken, J. W. M.2002,, 046101. doi: 10.1103/PhysRevLett.89.046101
(42) Ackermann, M. D.; Pedersen, T. M.; Hendriksen, B. L. M.; Robach, O.; Bobaru, S. C.; Popa, I.; Quiros, C.; Kim, H.; Hammer, B.; Ferrer, S.;.2005,, 255505. doi: 10.1103/PhysRevLett.95.255505
(43) Toyoshima, R.; Yoshida, M.; Monya, Y.; Suzuki, K.; Mun, B. S.; Amemiya, K.; Mase, K.; Kondoh, H.2012,, 3182. doi: 10.1021/jz301404n
(44) Butcher, D. R.; Grass, M. E.; Zeng, Z. H.; Aksoy, F.; Bluhm, H.; Li, W. X.; Mun, B. S.; Somorjai, G. A.; Liu, Z.2011,, 20319. doi: 10.1021/ja207261s
(45) Alayon, E. M. C.; Singh, J.; Nachtegaal, M.; Harfouche, M.; van Bokhoven, J. A.2009,, 228. doi: 10.1016/j.jcat.2009.02.010
(46) Chung, J. Y.; Aksoy, F.; Grass, M. E.; Kondoh, H.; Ross, J. P.; Liu, Z.; Mun, B. S.2009,, L35. doi: 10.1016/j.susc.2009.01.016
(47) Toyoshima, R.; Yoshida, M.; Monya, Y.; Suzuki, K.; Amemiya, K.; Mase, K.; Mun, BS.; Kondoh, H.2013,, 20617. doi: 10.1021/jp4054132
(48) Goodman, D. W.; Peden, C. H. F.; Chen, M. S.2007,, L124. doi: 10.1016/j.susc.2007.08.003
(49) McClure, S. M.; Goodman, D. W.2009,, 1. doi: 10.1016/j.cplett.2008.12.066
(50) Gao, F.; Cai, Y.; Gath, K. K.; Wang, Y.; Chen, M. S.; Guo, Q. L.; Goodman, D. W.2009,, 182. doi: 10.1021/jp8077979
(51) Huang, W. X.; Zhai, R. S.; Bao, X. H.2000,, 287. doi: 10.1016/S0169-4332(00)00010-6
(52) Zheng, G.; Altman, E. I.2002,, 253. doi: 10.1016/S0039-6028(02)01104-4
(53) Lundgren, E.; Gustafson, J.; Mikkelsen, A.; Andersen, J. N.; Stierle, A.; Dosch, H.; Todorova, M.; Rogal, J.; Reuter, K.; Scheffler, M.2004,, 46101. doi: 10.1103/PhysRevLett.92.046101
(54) Dumbuya, K.; Cabailh, G.; Lazzari, R.; Jupille, J.; Ringel, L.; Pistor, M.; Lytken, O.; Steinruck, H. P.; Gottfried, J. M.2012,, 20. doi: 10.1016/j.cattod.2011.09.035
(55) Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N.1987,, 405. doi: 10.1246/cl.1987.405
(56) Haruta, M.; Date, M.2001,, 427. doi: 10.1016/S0926-860X(01)00847-X
(57) Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M. J.; Delmon, B.1993,, 175. doi: 10.1006/jcat.1993.1322
(58) Haruta, M.1997,, 153. doi: 10.1016/S0920-5861(96)00208-8
(59) Valden, M.; Lai, X.; Goodman, D. W.1998,, 1647. doi: 10.1126/science.281.5383.1647
(60) Qiao, B. T.; Liang, J. X.; Wang, A. Q.; Liu, J. Y.; Zhang, T.2016,, 1580. [乔波涛, 梁锦霞, 王爱琴, 刘景月, 张涛. 催化学报, 2016,, 1580.] doi: 10.1016/S1872-2067(16)62529-9
(61) Chen, M. S.; Goodman, D. W.2005,, 259. doi: 10.1016/j.susc.2004.10.036
(62) Chen, M. S.; Cai, Y.; Yan, Z.; Goodman, D. W.2006,, 341. doi: 10.1021/ja0557536
(63) Chen, M. S.; Goodman, D. W.2006,, 739. doi: 10.1021/ar040309d
(64) Chen, M. S.; Goodman, D. W.2007,, 41. doi: 10.1007/s11244-007-0276-x
(65) Chen, M. S.; Luo, K.; Kumar, D.; Wallace, W. T.; Yi, C. W.; Gath, K. K.; Goodman, D. W.2007,, 632. doi: 10.1016/j.susc.2006.10.042
(66) Chen, M. S.; Goodman, D. W.2008,, 1860. doi: 10.1039/b707318f
(67) Kung, M. C.; Davis, R. J.; Kung, H. H.2007,, 11767. doi: 10.1021/jp072102i
(68) Wang, Z. W.; Wang, X. V.; Zeng, D. Y.; Chen, M. S.; Wan, H. L.2011,, 144. doi: 10.1016/j.cattod.2010.07.006
(69) Li, X. K.; Ma, D. D.; Zheng, Y. P.; Zhang, H.; Ding, D.; Chen, M. S. Wan, H. L.2015,, 1753. [李晓坤, 马冬冬, 郑燕萍, 张宏, 丁丁, 陈明树, 万惠霖. 物理化学学报, 2015,, 1753.] doi: 10.3866/PKU.WHXB201507091
(70) Zheng, W. W.; Chen, M. S. Lin, W. H.2002,, 102.
(71) Wei, J.; Iglesia, E.2004,, 4094. doi: 10.1021/jp036985z
(72) Hicks, R. F.; Qi, H.; Young, M. L.; Lee, R. G.1990,, 295. doi: 10.1016/0021-9517(90)90283-P
(73) Chin, Y. H.; Buda, C.; Neurock, M.; Iglesia, E.2011,, 15958. doi: 10.1021/ja202411v
(74) Weng, W. Z.; Chen, M. S.; Yan, Q. G.; Wu, T. H.; Chao, Z. S.; Liao, Y. Y.; Wan, H. L.2000,, 317. doi: 10.1016/S0920-5861(00)00475-2
(75) Weng, X. F.; Ren, H. J.; Chen, M. S.; Wan, H. L.2014,, 2598. doi: 10.1021/cs500510x
(76) Ciuparu, D.; Altman, E.; Pfefferle, L.2001,, 64. doi: 10.1006/jcat.2001.3331
(77) Monteiro, R. S.; Zemlyanov, D.; Storey, J. M.; Ribeiro, F. H.2001,, 291. doi: 10.1006/jcat.2001.3176
(78) Chen, M. S.; Weng, W. Z.; Wan, H. L.2000,, 6.
(79) Chen, M. S.; Weng, W. Z.; Wan, H. L.1998,, 542.
(80) Chen, M. S.; Weng, W. Z.; Wan, H. L.1999,, 938. [陈明树, 翁维正, 万惠霖. 物理化学学报, 1999,, 938.] doi: 10.3866/PKU.WHXB19991014
(81) Chen, M. S.; Weng, W. Z.; Wan, H. L.; Xu, P. P.1999,, 556.
(82) Avila, M. S.; Vignatti, C. I.; Apesteguia, C. R.; Rao, V. V.; Chary, K.; Garetto, T. F.2010,, 118. doi: 10.1007/s10562-009-0204-8
(83) Yazawa, Y.; Yoshida, H.; Komai, S.; Hattori, T.2002,, 113. doi: 10.1016/S0926-860X(02)00129-1
(84) Yao, Y. F. Y.. 1980,, 293.
(85) Garetto, T. F.; Rincón, E.; Apesteguı́a, C. R.2004,, 167. doi: 10.1016/j.apcatb.2003.10.004
(86) Tang, Z. Y.; Wang, S. L.; Zhang, L. L.; Ding, D.; Chen, M. S.; Wan, H. L.2013,, 12124. doi: 10.1039/c3cp50712b
(87) Lin, Y.; Xu, C. Y.; Chen, M. S.2013,, 68. [林瑛, 徐超毅, 陈明树. 厦门大学学报(自然科学版). 2013,, 68.]
(88) Kao, C.-L.; Madix, R. J.2002,, 8248. doi: 10.1021/jp020706a .
(89) Min, E. Z.; Du, Z. X.2002,, 1. [闵恩泽, 杜泽学. 当代石油石化. 2002,, 1.]
(90) Huang, J. J.; Song,Y. Y.; Ma, D. D.; Zheng, Y. P.; Chen, M. S.; Wan, H. L.2017,, 1229. doi: 10.1016/S1872-2067(17)62857-2
(91) Hu, J.; Song, Y. Y.; Huang, J. J.; Li, Y. Y.; Chen, M. S.; Wan, H. L.2017. doi: 10.1002/chem.201701697
(92) Han, Y. F.; Wang, J. H.; Kumar, D.; Yan, Z.; Goodman, D. W.2005,, 467. doi: 10.1016/j.jcat.2005.04.001
(93) Chen, M. S.; Goodman, D. W.2008,, 1178.
(94) Chusuei, C.C.; Lai, X.F.; Luo, K.; Guo, Q. L.; Goodman, D.W. Preparation of Thin-Film Alumina for Catalytic Activity Studies. In; Soriaga M. P. Eds. Springer: NY, USA, 2002; p. 253.
(95) Chen, M. S.; Santra, A. K.; Goodman D. W.2004,, 155404. doi: 10.1103/PhysRevB.69.155404
(96) Guo, Q.; Oh, W. S.; Goodman D. W.1999,, 49. doi: 10.1016/S0039-6028(99)00678-0
(97) Meriaudeau, P.; Naccache, C.1997,, 5. doi: 10.1080/01614949708006467
(98) Lundwall, M. J.; McClure, S. M.; Goodman, D. W.2010,, 7904. doi: 10.1021/jp9119292
(99) Lundwall, M. J.; McClure, S. M.; Wang, W. X.; Wang, Z. J.; Chen, M. S.; Goodman, D.2012,, 18155. doi: 10.1021/jp301824c
(100) Gillespie, W. D.; Herz, R. K.; Petersen, E. E.; Somorjai, G. A.1981,, 147. doi: 10.1016/0021-9517(81)90324-9
Toward Understanding the Nature of the Active Sites and Structure-Activity Relationships of Heterogeneous Catalysts by Model Catalysis Studies
CHEN Ming-Shu*
()
Understanding the nature of the active sites and the relationship between the catalyst structure and its performance are fundamental aspects of heterogeneous catalysis. With the development of modern surface science techniques, atomically resolved surface structures of heterogeneous catalysts and their properties can be studied with ease. Combined with anhigh pressure cell, model catalysis studies can provide convincing information about the relationship between the catalyst structure and its performance. In this mini-review, several case studies of model catalysts have been summarized, including those of the active surfaces for CO and alkane oxidation using the Pt group metals as catalysts, the active site of gold nanoparticles for CO oxidation, synergistic effects between VOand Pt for propane oxidation, promotional effects of Au in Pd-Au catalysts for vinyl acetate synthesis, structure−sensitivity of‑heptane dehydrocyclization on model oxide-supported Pt, as well as several significant improvements of the model catalysis techniques.
Model catalysis study; Structure-activity; CO oxidation; Alkanes oxidation;spectroscopy
June 17, 2017;
July 5, 2017;
July 17, 2017.
Corresponding author. Email: chenms@xmu.edu.cn; Tel: +86-592-2183723.
10.3866/PKU.WHXB201707171
O643
The project was supported by the National Basic Research Program of China (973 program: 2013CB933102) and National Natural Science Foundation of China (21273178, 21573180, 91545204).
973重大研究计划(2013CB933102)和国家自然科学基金(21273178, 21573180, 91545204).
猜你喜欢
陶瓷学报(2020年6期)2021-01-26
中学生数理化·高一版(2020年6期)2020-07-25
石油石化绿色低碳(2019年6期)2019-02-13
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
中学生数理化·八年级物理人教版(2017年3期)2017-11-09
信息安全与通信保密(2016年3期)2016-08-23
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
