
一种只利用序列信息预测RNA结合蛋白的深度学习模型
2018-01-12 07:19李洪顺宫秀军
计算机研究与发展 2018年1期
李洪顺 于 华 宫秀军
(天津大学计算机科学与技术学院 天津 300072)
(天津市认知计算与应用重点实验室(天津大学) 天津 300072)
(hongshunlee@foxmail.com)
RNA结合蛋白在选择性剪贴、RNA编辑及甲基化等多种生物功能中发挥非常重要的作用,从氨基酸序列预测这些蛋白成为基因组功能注释领域的主要挑战之一,只利用序列信息预测蛋白质功能一直是生物信息研究的热点问题.当前基于序列的计算方法主要包括基于序列同源性比对预测方法和基于特征提取的机器学习方法.基于序列同源性的方法比较典型的如BLAST[1], FASTA[2-3], PSI-BLAST[4].这些方法存在的主要问题是有些高同源性的蛋白质却表现着不同的功能[5].基于特征提取的方法主要从序列信息结合氨基酸的理化特性构造特征集,再利用机器学习方法进行功能预测[6-7].Cai等人[8]提出了从序列中提取氨基酸组成、范德华力、电荷、氨基酸疏水性、表面张力、极性、溶剂性、极化率和二级结构,并利用支持向量机模型进行预测.Wang等人[9-10]提出基于n-gram的特征构建方法,将提取出来的n-gram特征放入机器学习的分类器中.这种方法的问题在于当n的值变大时,提取出来的特征维度会呈指数型增长,计算量也会随之增长.为了减少n值过大带来的维度灾难,Gao等人[11]从序列中提取了56维特征,前20维特征是每种氨基酸在序列中出现的频率,之后将20种氨基酸按照生物化学特性分成6类,在这6类的基础上做2-gram,最终将提取出来的56维特征放到人工神经网络中对蛋白质进行分类.Shen等人[6]提出将氨基酸按照理化特性分为7类的特征提取方法,Lin等人[7]提出了n-gram和理化特性结合的188D方法.其他的预测蛋白质功能的方法还有基于蛋白质相互作用的方法[12]等.
然而上述计算方法存在诸多问题:1)特征提取方法没有提取出motif特征,motif与motif之间的位置信息也没有表示出来;2)基于序列同源性的计算方法将数据经过BLAST滤波,同源性高于某个阈值的序列要被移除,导致数据规模普遍太小,可信度不高;3)对噪声氨基酸{B,J,U,X,Z}没有有效的解决方案,但是在现实的数据中噪声数据是不可避免的.
2006年以来,“深度学习[13]”作为机器学习的新生课题开始受到学术界和工业界的广泛关注,已经成为人工智能和大数据的一个热潮[14].深度学习模拟人脑中分层的模型结构,数据被表示为一个具有多层的特征.目前深度学习在计算机视觉[15-16]、语音识别[17]领域取得了非常好的效果,并且在自然语言和生物信息[18]领域也逐渐崭露头角.
本文提出了一种从序列预测RNA结合蛋白的深度学习模型,实验所用到的特征都是通过“学习”自动获得.模型利用2阶段卷积神经网络探测蛋白质序列的功能域,利用长短期记忆神经网络对功能域之间的长期依赖关系进行学习.在实验中使用的数据都是原始的序列数据,序列数量达到10万级,在测试集上的精度达到0.959 2,实验结果表明卷积神经网络和长短期记忆神经网络结合能够有效地预测蛋白质是否含有与RNA结合的功能.
1 模型描述
一条蛋白质序列可以描述为s=a1a2…an,其中n为蛋白质序列的长度,ai来自氨基酸的集合(包括噪声蛋白),∑={A,B,…,Z},为了预测一条蛋白质序列是否具有RNA-Binding功能,设计了一个有6个阶段组成的深度学习模型,其工作流程如图1所示.
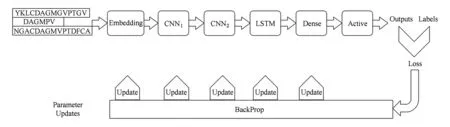
Fig. 1 The workflow of model图1 模型的工作流程
1.1 数据预处理
假设数据集D包含m个样本,D={(s1,y1),(s2,y2),…,(sm,ym)}.其中si为蛋白质序列;yi⊆{0,1}表示蛋白质功能标签,0代表没有RNA-Binding功能,1代表有RNA-Binding功能;si=a1a2…an,ai为氨基酸,n为si的长度.为了使蛋白质序列能够被计算机识别,工作的第1步就是对蛋白质中的每个氨基酸根据表1进行编码,将每个氨基酸映射成为一个具体的实数.

Table 1 The Amino Acids Encoder表1 氨基酸编码对照表
图1中的Embedding阶段,将1维向量中的各个实数映射为固定长度的1维向量;CNN是卷积操作,它首先利用多个卷积核去对矩阵进行卷积操作,卷积出来的卷积特征可以理解为蛋白质的功能域特征;池化操作对卷积出来的特征的一部分区域取均值或者最大值,这里选取的是最大值,从而得到卷积阶段的特征映像,而特征映像与特征映像之间的关系可以理解为蛋白质功能域之间的位置信息;LSTM是循环神经网络RNN的一种性能更好的模型,它将由卷积阶段产生的特征映像以时间步长的形式输入到LSTM神经网络中,输出一个固定长度的向量;Dense操作是将LSTM输出的固定长度的向量映射为一个具体的数字;Active是激活层,选取的是sigmoid函数,目的是将输出映射到[0,1]区间,从而得到一个预测的置信值.在反向传播过程中,模型会根据输出的Outputs和真实的Labels之间的差距算出一个损失,根据损失依次算出Dense阶段、LSTM阶段、CNN阶段和Embedding阶段中参数的梯度,用计算出来的梯度更新各个阶段的参数,从而完成一次学习过程.
首先对氨基酸按照字母的顺序从小到大进行编码,表1只是单纯地为了方便起见,编码的顺序不影响实验结果.
假设一条长度为7的序列s=MSFVPT,首先根据表1进行编码,将序列就转化为一个1维向量.
S1=encoding(s)=(11,16,5,11,18,13,17).
重构的过程是将得到的1维向量变成等长过程,不足的地方填充为0,这里我们假设maxlength=8,所以重构之后的向量为
S2=(0,11,16,5,11,18,13,17).
1.2 嵌入(embedding stage)阶段
嵌入阶段会将向量中的每一个数字映像成为一个固定长度的向量,这里为了方便,假设fixedlength=8,这样在经过嵌入阶段之后,上一阶段得到的向量就变成了一个8×8的矩阵:

1.3 卷积(CNN stage)阶段
卷积阶段会利用多个卷积核W去探测这些矩阵,其中W∈in×k,k是Embedding阶段设置的fixedlength,
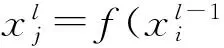
(1)
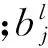
RELU(x)=max(0,x).
(2)
该激活函数相比函数sigmoid具有便于稀疏化及有效减少梯度似然值的优势.
卷积操作的主要工作流程如图2所示:
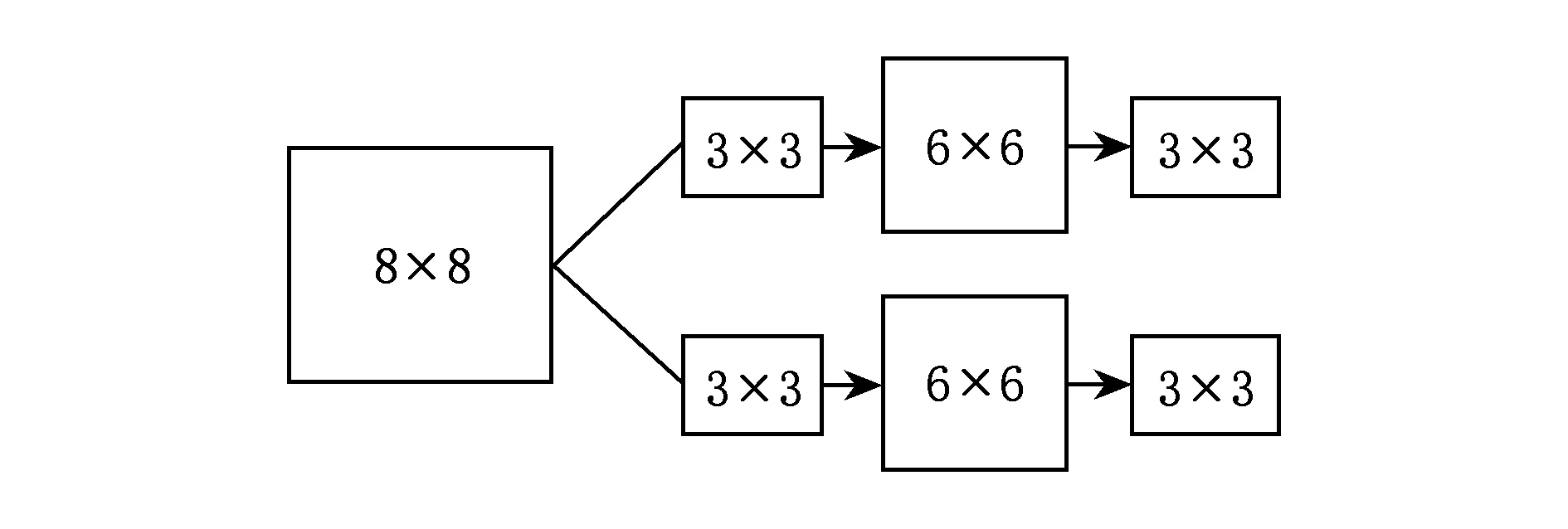
Fig. 2 The structure of CNN图2 卷积神经网络的结构图
图2中首先利用2个3×3的卷积核去探测初始的8×8的方阵从而得到了2个6×6的方阵;接着对其进行下采样最终得到了2个3×3的特征映射.这里为了更直观地介绍卷积操作,我们假设用1个2×8的卷积核:
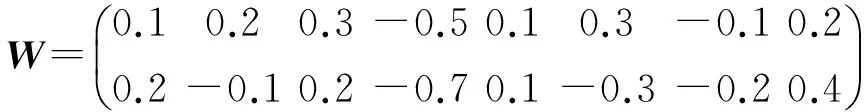
去探测由嵌入阶段产生的结果:
最后的结果S4称之为由卷积核W探测出来的特征映像.本实验设计了2个卷积层,每层都是用64个卷积核去卷积,第1层用的是10×20的卷积核,第2层用的是长度为5×1的卷积核,这样4 096个特征映像就被提取出来.
1.4 长短期记忆神经网络阶段(LSTM stage)
长短期记忆神经网络[19],它主要用1个特定的记忆细胞去存储信息,对于存在依赖关系的资料有着非常显著的性能,LSTM对于处理变长序列的效果是非常明显的[20],目前LSTM已经成功应用到语音识别和手写体识别领域[17,21],LSTM的记忆细胞结构如图3所示[19]:
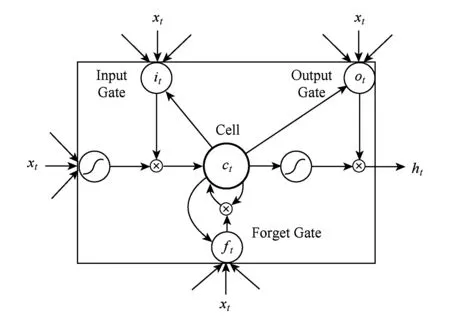
Fig. 3 The memory cell structure of LSTM图3 长短期记忆神经网络记忆细胞结构
(3)
ft=σ(Wx fxt+Wh fht-1+Wcfct-1+bf),
(4)
ct=ftct-1+ittanh(Wx cxt+Wh cht-1+bc),
(5)
ot=σ(Wx oxt+Wh oht-1+Wc oct+bo),
(6)
ht=ottanh(ct),
(7)
其中,σ是sigmoid函数;i,f,o,c分别代表输入门、遗忘门、输出门、记忆细胞和记忆细胞的启动向量.它们作为隐藏层h有着同样大小的规模.从权值矩阵的下标就能看出每一个权值矩阵的具体含义,例如Wh i代表的就是隐藏输入门的权值矩阵.LSTM会产生固定长度的特征表示:
S5=LSTM(S4)=(a1,a2,…,an),
其中,这样一条序列经过上述5个阶段就生成了一个固定长度为n的向量,也可以称之为学习出来的特征表示.
1.5 密集化阶段(dense stage)
密集化阶段的主要功能是将由LSTM层产生的固定长度的向量转化为一个特定长度的向量.
S6=dense(S5,n)=(a1,a2,…,an),
其中,n为经过Dense后输出的数字个数,这里我们取n=1,这样就将LSTM层输出的特征映像成为一个具体的数字,即S6是一个具体的实数.
1.6 激活阶段(active stage)
模型的最后一层为启动层,采用的启动函数为sigmoid函数,它能将一个值映射到[0,1]区间,从而完成预测.
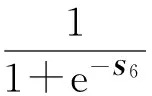
最后算出预测值与真实值之间的损失,损失函数使用的是根据这个损失逐层地去计算参数对应的梯度,根据梯度更新参数,完成一次学习的过程.
1.7 模型实现
我们的模型是用Keras框架设计并实现的,Keras框架是一个基于Theano、并用Python语言编写的高度模块化的深度学习框架,它的设计参考了Torch,并且能够同时支持CPU和GPU.
模型嵌入阶段的参数选择.嵌入阶段主要有3个参数:输入维度input_dim=23、输出维度output_dim=128、输入的长度input_length=1 000.在第1个卷积阶段我们使用64个10×20的卷积核去卷积;第2个卷积阶段除了卷积核变为5×20之外与第1个卷积阶段一样,长短期记忆神经网络阶段只有1个参数output_length=70;紧接着LSTM阶段的是Dense层,它只有1个参数设为1,即将LSTM输出的长度为70的1维向量密集化一个具体的数;最后1层为启动层,它的参数为‘sigmoid’,即用sigmoid函数去启动,从而得到预测的置信值,实验中的模型我们加入了1个dropout层,参数设为0.5,损失函数loss=‘binary_crossentropy’,batch_size=128,迭代次数设为30.实验用到的部分数据和代码已经上传至GitHub*https://github.com/lihongshun/bioinformation-lab.
2 实验结果
2.1 数据选择
本实验所用到的训练数据是从Uniprot数据库中选择经过人工注释和修订过的Swiss-prot数据库,总共有551 193条蛋白质序列,之后使用关键词“RNA-Binding”进行搜索,并且加上限制条件:序列的长度要介于40~1 000之间,最终得到了55 881条蛋白质序列,并将这些数据作为正例数据;在剩下的资料中添加限制条件:序列长度介于40~1 000之间,并且该序列有着分子功能,从剩下的数据中随机选取了56 081条序列作为反例;为了验证模型的泛化能力,从PDB数据库中用同样的方法选取了一部分数据作为验证集,如表2所示.我们称该数据集为类别平衡数据集,学习到的模型称为类别平衡模型.
然而现实的生物数据中蛋白质序列中含有RNA-Binding功能的蛋白质序列只占所有序列的小部分,所以基于现实数据的考虑我们又构建了一个类别非平衡数据集,学习到的模型称为类别非平衡模型,如表3所示.
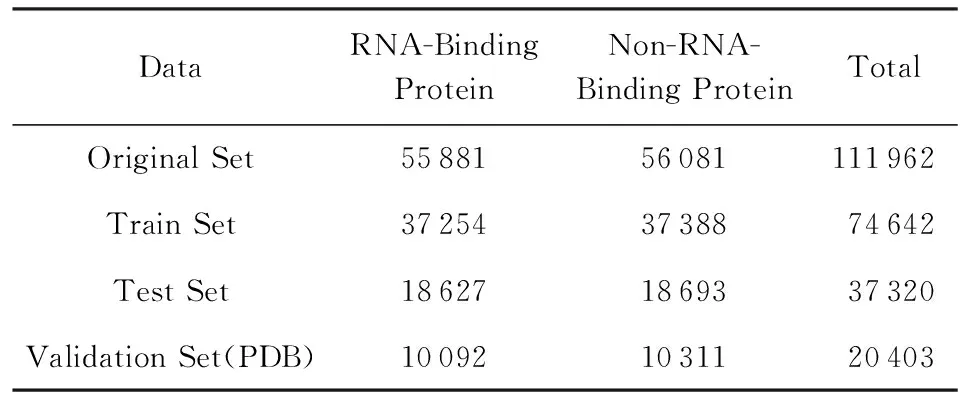
Table 2 Equal Data Set表2 平衡数据集

Table 3 Unbalance Data Set表3 非平衡数据集
在平衡数据集中,我们随机地选取66.7%的数据作为训练数据、33.3%的数据作为测试数据,在非平衡数据集中选取了80%的数据作为训练数据、20%的作为测试资料.
2.2 传统的特征提取方法
1) 三元组法(conjoint triad, CT).在Shen等人[6]的研究中,一条蛋白质序列会根据氨基酸的理化特性将蛋白质分成7个类别:A={A,G,V},B={C},C={D,E},D={F,I,L,P},E={H,N,Q,W},F={K,R},G={M,S,T,Y}.三元组法计算分类好的氨基酸集合的3-gram,即计算集合Σ={AAA,AAB,…,GGG}中每个元素出现的频率,这样一个343维的特征就被提取出来了.
2) 188D.Lin等人[7]提出了从序列信息提取188维特征的特征提取方法,其中前20维是每种氨基酸出现的频率,后面的168维是根据每种氨基酸的理化特性提取出来的.
2.3 在平衡数据集下的结果
本次实验在平衡数据集上采用3次交叉验证法来验证模型的性能,3次交叉验证的思想是将数据集等分为3个互不相交的数据集,每次取其中的一个作为验证集,其余的2个作为训练集:
D=D1∪D2∪D3,
其中,Di∩Dj=∅,i≠j.3次交叉验证后的平均精度为0.959 2,说明我们的模型具有一定稳定性和保真性,图4展示了3次交叉验证的结果.

Fig. 4 The result of 3-fold-cross validation图4 3次交叉验证的结果
为了验证模型的泛化能力,实验从PDB数据库中用关键词RNA-Binding作为关键词搜索,并且添加限制条件序列长度要大于40小于1 000,这样10 092条数据就被得到作为正例数据,从剩下的数据中随机选取10 311条序列作为反例构造验证集.结果如表4所示:

Table 4 The Results in Test Data Set and Validation Data Set表4 在测试集和验证集上的结果
实验结果表明,在非同源的数据上模型也具备一定的泛化能力.
2.4 与其他特征提取方法的对比
为了验证深度学习模型在大规模数据集上的优越性,实验选取了2个传统的特征提取方法,并用支持向量机和逻辑回归分别对三元组法和188D方法进行实验,数据用到的是表2的原始数据,其中80%的数据作为训练集,其余的20%作为测试集,评价指标分别用:
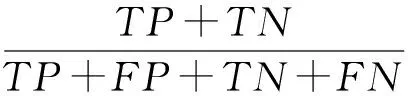
其中,TP表示本身是正例,预测结果也是正例的个数;FN表示本身是正例,但是分类器将其判定为反例的个数;FP表示本来是反例,但是分类器将其判定为正例的个数;TN表示将本身就是反例预测结果也是反例的个数.结果如表5所示:
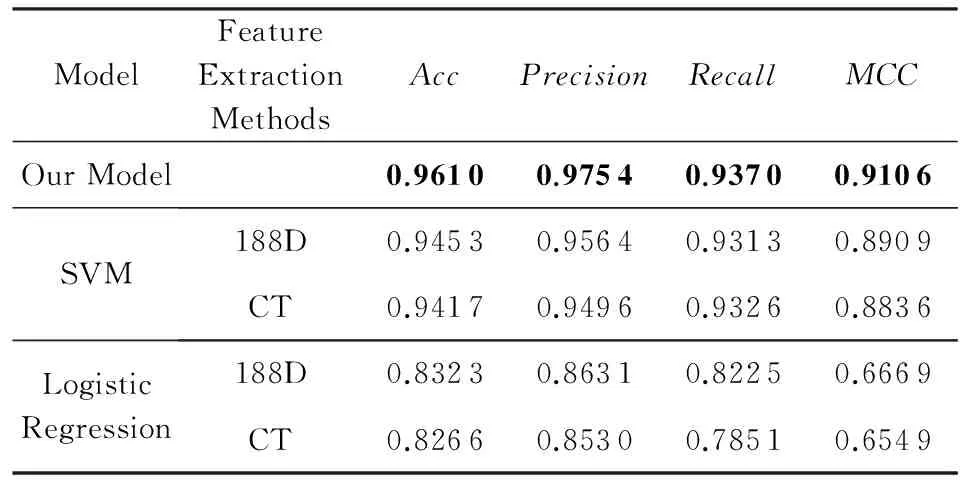
Table 5 The Results in Different Models and Different Feature Extraction Methods
从实验结果可以看出,我们设计的深度学习模型来处理蛋白质数据在大规模数据下比以往的特征提取方法结合机器学习方法有一定的优势.
2.5 在非平衡数据集下的实验结果
在真实的生物系统中与RNA结合的蛋白只占所有蛋白质的一小部分.我们根据现实情况构造了一个非平衡模型,之后在PDB数据库中选择了一组非平衡的数据并且加限制条件“长度大于40小于1 000”作为验证集.因为数据是非平衡的,所以单纯的精度不能表明模型的预测能力,这里我们采用Sensitive,Specificity,Auc来评价模型的性能,其中:
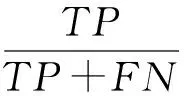
表示在预测结果中预测正确的正例占所有正例的比例;
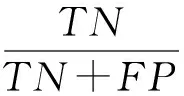
表示在预测结果中预测正确的反例占所有反例的比例.结果如表6所示:

Table 6 The Results in Unbalance Data Set表6 模型在非平衡数据下的结果
Keras框架既支持{0,1}这样的二进制输出,同时也支持概率输出,所以相应的ROC曲线也能得到,如图5~6所示.
图5展示了在测试集上的ROC曲线,从结果可以看出在测试集上的ROC曲线已经非常接近真实情况.
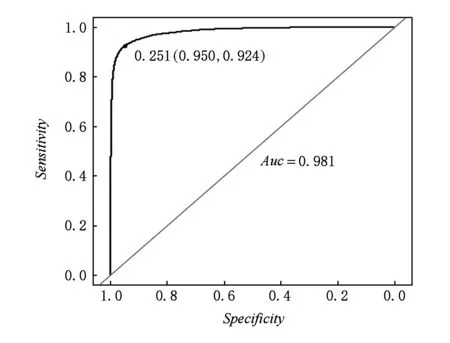
Fig. 5 The ROC of test data set on unbalance model图5 非平衡模型在测试集上的ROC曲线
图6展示了在验证集上的ROC曲线,结果显示,在不同源的数据集上模型的效果也是非常显著.

Fig. 6 The ROC of validation data set on unbalance model图6 非平衡模型在验证集上的ROC曲线
3 讨 论
本文中我们提出了一种基于深度学习的模型去预测一条蛋白质是否含有RNA结合的分子功能.深度学习也叫做特征学习,它能从高吞吐量的数据中学习出一组能够结构化表示以前数据的特征,我们主要用到的是卷积神经网络和长短记忆神经网络,卷积神经网络利用卷积核的局部感知去探测序列中比较重要的一小段子序列,长短期记忆神经网络将这些探测出来的子序列之间的依赖关系学习出来,而其中LSTM尤为重要,二者的结合将成为处理文本信息的新的研究方向.最后,0.959 2的精度说明我们的模型有着非常显著的效果,之后从PDB数据库中选取了一部分作为验证集,PDB数据库与Uniprot数据库属于非同源的数据库,从实验结果可以看出也得到了不错的效果,说明模型有较强的泛化能力.
在计算机视觉领域,已经有很多实验[17]表明卷积神经网络的深度对实验结果的影响很大,例如,由斯坦福大学提出的ImageNet竞赛中,从CNN被提出到最近网络的模型更是从16层[22]到30层[23],2015年第1名的队伍更是大胆地提出了152层的深度网络[24],结果已经超过了人类专家对图像认知的精准度.因此在实验阶段我们设计了2个不同的模型,第1个模型与本实验中的模型网络结构相同,第1个卷积层的卷积核长度不同,第2个模型相比于本实验中的模型少了一个卷积层;接着分别从损失的收敛速度和精度的提升速度做了对比.图7是在25次迭代中损失的收敛速度对比,图8是25次迭代中精度提升的速度对比,其中CNN( )的括号中的数字表示卷积核的个数.
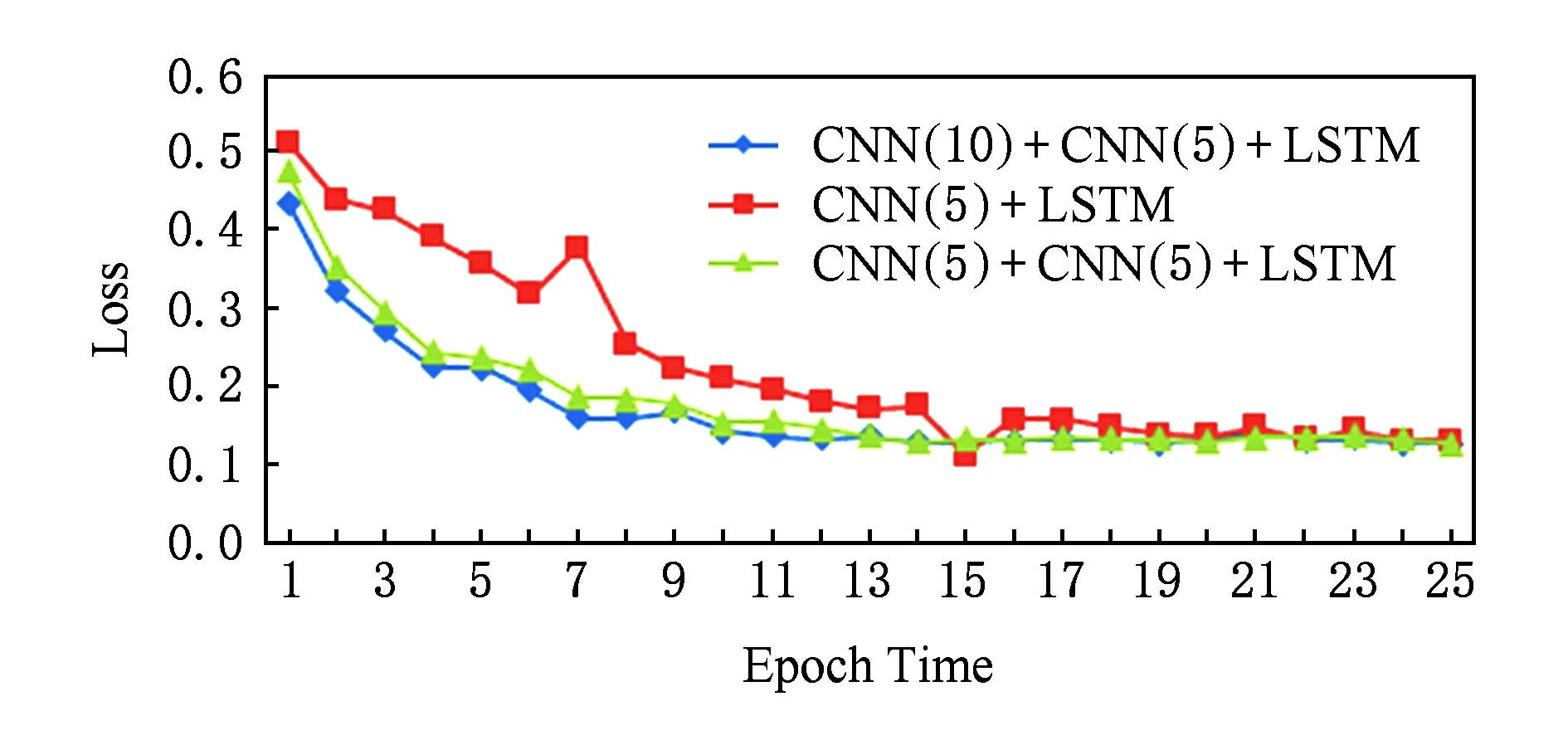
Fig. 7 Loss comparison in different models图7 不同模型loss收敛速度对比
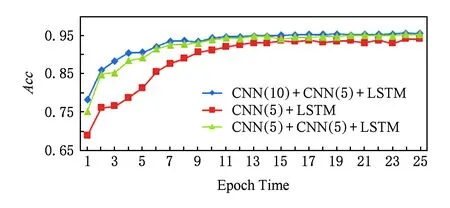
Fig. 8 Acc comparison in different models图8 不同模型精度提升的速度对比
结果表明:随着网络结构的复杂化,收敛速度和精度都有了一定的提升.
用卷积神经网络和长短期记忆神经网络来处理生物序列信息可以有效地利用卷积神经网络中局部感知的优势去探测序列中的motif信息,这样既发挥了CNN的优势又充分契合了生物序列中含有motif的生物意义.而长短期记忆神经网络可以将motif与motif之间的依赖关系学习出来,将2者结合能够很有效地处理类似的序列信息.
4 总结与展望
在实验过程,我们发现数据的选择往往比人工的去设计模型更有效,一组好的数据往往能通过深度学模型学到更具规律性的特征,这就意味着,随着蛋白质序列数据的日益增加,这些高质量的数据会更容易得到,而随着数据量的增加深度学习模型的优势也会越来越显现出来.
在接下来的研究中,我们会将传统的机器学习方法与深度学习相结合发挥各自的优点,使深度学习学习出来的特征能够在传统的机器学习中发挥更重要的价值.
在未来的工作中我们将会尝试从更多的数据库中选取数据,而不是单一地从Uniprot中选择,这样数据的规模将更大,学习的效果也会有进一步的提升.未来我们还将优化模型及参数,也会试图将模型构建得更深,这样在不发生梯度消失的情况下,原始数据将会被更加结构化地表示.
[1] Moore K L. The biology and enzymology of protein tyrosine O-sulfation[J]. Journal of Biological Chemistry, 2003, 278(27): 24243-24246
[2] Shen Niu, Huang Tao, Feng Kaiyan, et a1. Prediction of tyrosine sulfation with mRMR feature selection and analysis[J]. Journal of Proteome Research, 2010, 9(12): 6490-6497
[3] Radivojac P, Vacic V, Haynes C, et al. Identification, analysis, and prediction of protein ubiquitination sites[J]. Proteins-Structure Function and Bioinformatics, 2010, 78(2): 365-380
[4] Gnad F, Ren Shubin, Choudhary C, et a1. Predicting post-translational lysine acetylation using support vector machines[J]. Bioinformatics, 2010, 26(13): 1666-1668
[5] Gnad F, Ren Shubin, Cox J, et al. PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites[J]. Genome Biology, 2007, 8(11): R250
[6] Shen Juwen, Zhang Jian, Luo Xiaomin, et al. Predicting protein-protein interactions based only on sequences information[J]. Proceedings of the National Academy of Sciences, 2007, 104(11): 4337-4341
[7] Lin Chen, Zou Ying, Qin Ji, et al. Hierarchical classification of protein folds using a novel ensemble classifier[J]. PloS One, 2013, 8(2): e56499
[8] Cai C Z, Han L Y, Ji Z L, et al. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence[J]. Nucleic Acids Research, 2003, 31(13): 3692-3697
[9] Wang Dianhui, Lee N K, Dillon T S, et al. Protein sequences classification using radial basis function (RBF) neural networks[C] //Proc of Int Conf on Neural Information. Piscataway, NJ: IEEE, 2002: 764-768
[10] Wang Dianhui, Lee N K, Dillon T S. Extraction and optimization of fuzzy protein sequences classification rules using GRBF neural networks[J]. Neural Information Processing-Letters and Reviews, 2003, 1(1): 53-57
[11] Gao J, Xiong L. Protein sequence classification with improve extreme learning machine algorithms[OL]. 2014 [2016-05-06]. https://www.hindawi.com/journals/bmri/2014/103054/abs
[12] Chua H N, Sung W K, Wong L. Exploiting indirect neighbours and topological weight to predict protein function from protein-protein interactions[J]. Bioinformatics, 2006, 22(3): 1623-1630
[13] Hinton G E, Osindero S, Teh Y W. A fast learning algorithm for deep belief nets[J]. Neural Computation, 2006, 18(7): 1527-1554
[14] Yu kai, Jia Lei, Chen Yuqiang, et al. Deep learning: Yesterday, today, and tomorrow[J]. Journal of Computer Research and Development, 2013, 50(9): 1799-1804 (in Chinese)
(余凯, 贾磊, 陈雨强, 等. 深度学习的昨天、今天和明天[J]. 计算机研究与发展, 2013, 50(9): 1799-1804)
[15] Krizhevsky A, Sutskever I, Hinton G E. Imagenet classification with deep convolutional neural networks[C] //Advances in Neural Information Processing Systems. Cambridge, MA: MIT Press, 2012: 1097-1105
[16] Lü Qi,Dou Yong, Niu Xin, et al. Remote sensing image classification based on DBN model[J]. Journal of Computer Research and Development, 2014, 51(9): 1911-1918 (in Chinese)
(吕启, 窦勇, 牛新, 等. 基于DBN模型的遥感图像分类[J]. 计算机研究与发展, 2014, 51(9): 1911-1918)
[17] Graves A, Mohamed A, Hinton G. Speech recognition with deep recurrent neural networks[C] //Proc of Int Conf on Acoustics, Speech and Signal Processing. Piscataway, NJ: IEEE, 2013: 6645-6649
[18] Alipanahi B, Delong A, Weirauch M T, et al. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning[J]. Nature Biotechnology, 2015, 33(8): 831-838
[19] Hochreiter S, Schmidhuber J. Long short-term memory[J]. Neural Computation, 1997, 9(8): 1735-1780
[20] Sutskever I, Vinyals O, Le Q V. Sequence to sequence learning with neural networks[C] //Advances in Neural Information Processing Systems. Cambridge, MA: MIT Press, 2014: 3104-3112
[21] Graves A, Schmidhuber J. Offline handwriting recognition with multidimensional recurrent neural networks[C] //Advances in Neural Information Processing Systems. Cambridge, MA: MIT Press, 2009: 545-552
[22] Simonyan K, Zisserman A. Very deep convolutional networks for large-scale image recognition[EB/OL]. (2014-09-04) [2016-06-01]. https://arxiv.org/abs/1409.1556
[23] Ioffe S, Szegedy C. Batch normalization: Accelerating deep network training by reducing internal covariate shift[EB/OL]. (2015-02-11) [2016-06-05]. https://arxiv.org/abs/1502.03167
[24] He K, Zhang X, Ren S, et al. Deep residual learning for image recognition[EB/OL]. (2015-12-10) [2017-01-10]. https://arxiv.org/abs/1512.03385
猜你喜欢
信阳农林学院学报(2022年2期)2022-08-06
新高考·高一数学(2022年3期)2022-04-28
北京航空航天大学学报(2021年9期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
湖南饲料(2021年3期)2021-07-28
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19
电子制作(2019年13期)2020-01-14
电子制作(2019年11期)2019-07-04
北京航空航天大学学报(2018年1期)2018-04-20
高中生学习·高三版(2016年9期)2016-05-14
