
儿童胱氨酸尿症临床及基因分析
2018-01-03 07:27:07马艳艳肖海雪刘玉鹏袁馥梅李东晓宋金青李溪远杨艳玲
临床儿科杂志 2017年12期
马艳艳 肖海雪 刘玉鹏 袁馥梅 李东晓 宋金青 李溪远 丁 圆 杨艳玲
1.青海大学附属医院(青海西宁 810001);2.北京大学第一医院儿科(北京 100034)
儿童胱氨酸尿症临床及基因分析
马艳艳1肖海雪1刘玉鹏2袁馥梅1李东晓2宋金青2李溪远2丁 圆2杨艳玲2
1.青海大学附属医院(青海西宁 810001);2.北京大学第一医院儿科(北京 100034)
目的 探讨以肾结石起病的胱氨酸尿症患儿的临床表现和基因突变。方法 回顾3例以肾结石起病的胱氨酸尿症患儿的临床资料,以及通过PCR扩增测序测定的SLC3A1和SLC7A9基因结果。结果 3例男性患儿来自三个无关家系,2例于1岁时、1例于14岁时因肾结石就诊。3例患儿的血氨基酸谱无异常,游离肉碱降低;尿氨基酸谱分析提示胱氨酸、鸟氨酸、精氨酸和苏氨酸部分增高。基因分析证实1例为SLC7A9基因c.325G>A纯合突变,父母为c.325G>A杂合突变的携带者;另2例均为SLC3A1基因复合杂合突变,分别为c.1365delG和c.1113C>A复合杂合突变,c.1897_1898insTA和c.1093C>T复合杂合突变,其父母均为杂合突变携带者。经确诊胱氨酸尿症后,予枸橼酸钾、左卡尼汀等治疗,患儿病情好转。结论 对于肾结石患儿应高度重视可能存在的遗传代谢病,尿液氨基酸分析、基因检测是确诊胱氨酸尿症的重要方法。
肾结石; 胱氨酸尿症; 氨基酸; 遗传代谢病
肾结石是肾脏疾病常见的临床症状之一,胱氨酸结石是肾结石的一种,如果结石阻塞尿道可引起肾功能不全。胱氨酸在酸性和中性尿液中溶解度降低,在尿道中易形成结晶或结石。通常情况下肾小球滤过液中99%的胱氨酸在近端肾小管刷状缘细胞被重吸收,当尿中胱氨酸浓度超过肾小球滤过负荷浓度的50%~200%或尿pH值降低时,胱氨酸可形成特有的六方结晶体[1]。胱氨酸尿症是一种常染色体隐性遗传病,因胱氨酸和一些二碱基氨基酸在近端肾小管或胃肠道重吸收和转运障碍导致的代谢障碍。1908年首次报道本病,随着生化诊断技术的提高,有关胱氨酸尿症的发病机制逐步明确,国内对成人胱氨酸结石患者的诊断与排石治疗研究较多[2-4],关于儿童胱氨酸尿症的临床表现及分子遗传学报道较少。现就3例胱氨酸尿症患儿的临床经过、生化和基因突变特点进行分析探讨。
1 临床资料
3例患儿来自不同的家庭,无亲缘关系,均为男性。例1,1岁时因腹泻就诊,查尿红细胞增多而行肾脏超声检查,发现双肾结石,尿液晶体检查存在典型的六角形胱氨酸结晶;2岁时复查发现结石较前增大,于腹腔镜下取石,此后服用甜菜碱和10%的枸橼酸钾维持治疗,目前患儿6岁,智力运动及体格发育正常,体力好,夜尿不多,无腹痛、尿痛、血尿等异常。全身皮肤无黄染、皮疹及出血点,心肺及腹部检查未见异常。例2,14岁时无明显诱因右侧腰痛2个月就诊,无尿痛,无发热、咳嗽和腹泻;肾B超示双肾髓质钙沉着,右肾充盈,输尿管、膀胱未见异常,诊断为肾结石;尿液晶体检查存在典型的六角形胱氨酸结晶,尿胱氨酸含量显著增高,经体外碎石治疗后好转,给予左卡尼汀1.0 g /d、甜菜碱1g /d、和10%的枸橼酸钾口服;6个月后复查,未发现新生结石,一般状况良好。例3,1岁时因排尿时哭闹3天就诊,夜哭,不愿排尿,无发热、腹泻和咳嗽,曾有10余小时不能排尿,并自行排出0.5mm的结石;经超声检查诊断为肾结石、尿道结石,经尿道手术取石;术后3个月复诊,左肾结石增大1.8 cm×0.2 cm,给予左卡尼汀1.0 g/d,甜菜碱1g/d和10%的枸橼酸钾,每日3次,每次10mL口服。患儿目前4岁,发育正常,肾结石无明显增大(表1)。
采用液相串联质谱法测定血液氨基酸及酯酰肉碱谱,定量分析采用软件ChemoView 1.2版本,根据同位素内标和各种丁酯化的氨基酸和酰基肉碱的离子峰强度计算标本中氨基酸和酰基肉碱浓度[5]。收集患儿新鲜尿液5~10mL,先测定尿肌酐浓度,分取相当于含0.2mg肌酐的尿样,萃取有机酸,采用气相色谱质谱联用分析仪进行有机酸测定[6]。3例患儿的血液总同型半胱氨酸均正常,鸟氨酸均降低,例2和例3的精氨酸降低,例2血苏氨酸和甲硫氨酸降低。3例患儿的血液游离肉碱均轻度降低,酯酰肉碱谱正常;尿液草酸、甘油酸等有机酸正常,胱氨酸、鸟氨酸、精氨酸和苏氨酸部分升高(表2)。

表 1 3例胱氨酸尿症患儿临床和生化特点
在知情同意的前提下,留取3例患儿及其父母静脉血,使用TIANGEN血液基因组DNA提取试剂盒(北京天根生化科技有限公司,生产批号DP318-03,离心柱型,200T,0.1~1 mL)提取外周血白细胞DNA。采用二代基因测序技术进行胱氨酸尿症相关基因分析。异常结果均经一代测序验证。
3例患儿分别检出SLC7A9或SLC3A1基因突变。例1为SLC7A9基因第4外显子c.325G>A纯合突变,突变导致中性的天冬酰胺被酪氨酸替代。其父母为c.325G>A突变携带者。例2和例3均检出SLC3A1基因复合杂合突变,例2为SLC3A1基因c.1365delG和c.1113C>A杂合突变,c.1365delG突变导致蛋白编码链提前终止,c.1113C>A突变导致第371变为终止密码;例3为SLC3A1基因c.1897_1898insTA和c.1093C>T杂合突变,这些突变导致相应蛋白质合成有不同形式改变(表3)。以上5种突变未在100例正常人群中和人类千人计划数据库中发现,均为新突变。
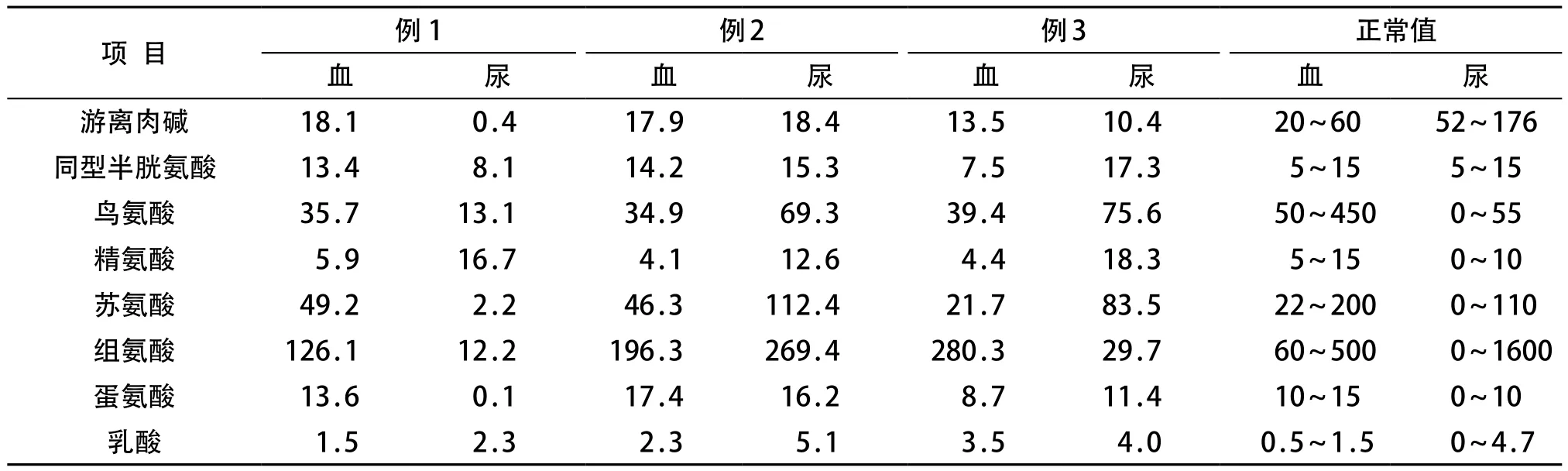
表 2 3例胱氨酸尿症患儿血和尿氨基酸浓度 (μmol/L)
2 讨论
胱氨酸尿症是一种常染色体隐性遗传病,由于近端肾小管上皮细胞对胱氨酸、赖氨酸、鸟氨酸和精氨酸转运障碍,使这些氨基酸不能被近端肾小管重吸收,在尿中随着氨基酸浓度的增高而影响肾功能。胱氨酸尿症患者具有显著的基因及表型异质性,临床症状复杂。胱氨酸在酸性和中性尿中溶解度降低,当尿液胱氨酸异常增高时可形成结晶或结石,损害肾小管功能。我国胱氨酸尿症发病率不详,美国约1∶15 000,瑞士约为1∶100 000[7]。
胱氨酸尿症患者可出现与肾结石相关的症状和体征,有时可表现为无肾损害的鹿角型结石[8]。25%的患者在10岁以内首次发病,30%~40%的患者于青少年期发现肾结石[9]。美国胱氨酸尿症患儿形成结石的中位年龄12岁,男性较女性更重[10]。有研究表明,40%的患者在11~20岁首次出现肾结石表现,14%的女性和28%的男性患者在3岁之前就已经有肾结石的症状[11]。本研究中3例患儿均为男童,2例于1岁、1例14岁发病,首次发病均为肾结石表现。
既往,根据携带杂合突变患者尿中胱氨酸的分泌量将胱氨酸尿症分为3个亚型,这种表型分类很难体现基因信息。目前多采用根据基因信息分类的方法,分为A型和B型。A型是指因SLC3A1基因突变致病的患者,B型是指因SLC7A9基因突变致病的患者。本研究中例1属于B型,例2和例3属于A型。
迄今,国外已报道SLC3A1基因130种和SLC7A9基因100种致病性突变,基因突变谱较广,包括无义、错义、剪切和框移突变[12]。其中在SLC7A9基因外显子5-9的缺失和重复突变较多见[13]。本组3例患儿及其父母检出了5种新突变,分别是SLC7A9基因c.325G>A突变,SLC3A1基因c.1365delG、c.1113C>A、c.1897_1898insTA和c.1093C>T。例1为B型,SLC7A9基因第4外显子c.325G>A纯合突变,父母均为c.325G>A杂合突变携带者,无肾结石,符合常染色体隐性遗传模式。但是,有研究显示,SLC7A9基因突变倾向于不完全显性遗传,即SLC7A9基因杂合突变携带者部分也可表现为肾结石[1]。SLC3A1基因突变为常染色体隐性遗传,本组例2、例3患儿为SLC3A1复合杂合突变,符合A型,其中一个为框移突变。例2的SLC3A1外显子8存在c.1365delG,外显子6检出c.1113C>A无义突变。例3的SLC3A1基因外显子10存在c.1897_1898insTA突变,外显子6存在c.1093C>T错义突变,提示SLC3A1外显子5-9的缺失和重复突变应该被列为胱氨酸尿症的基因筛查种类之一,与既往报道一致。
肾脏结石是儿科临床较为常见的疾病之一。尿中各种成分异常或比例失衡引起尿液中晶体形成,在与肾小管上皮细胞作用形成结石。引起尿成分异常的原因比较复杂,尿中草酸、钙盐、尿酸、枸橼酸、胱氨酸异常,均能导致肾结石。尤其对于除泌尿系统外还有其他一个或多个系统同时受累的患儿,应考虑遗传代谢病的可能。胱氨酸尿症是近年来临床逐渐被认识的遗传代谢病。对于表现为肾结石的患儿注意排除胱氨酸尿症。早期诊断和治疗可防止或减缓结石的反复出现,保护肾脏功能。
胱氨酸尿症治疗困难,胱氨酸结石术后极易复发、体外冲击波碎石术常不能将其粉碎、经皮肾镜取石术后残余结石率较高,故胱氨酸结石的长期治疗是一个非常棘手的问题[14,15]。胱氨酸尿症治疗的主要目标是降低尿中胱氨酸浓度,提高胱氨酸在尿中的溶解度,促进排泄。主要治疗方法为鼓励饮水,增加液体摄入,补充碱性药物,提高尿液pH值,以减少胱氨酸排泄,增加胱氨酸溶解度[16]。由于胱氨酸及其前体物质甲硫氨酸的摄入量与尿胱氨酸的分泌呈正相关,同时动物蛋白是质子的重要来源,蛋白摄入增多降低尿pH值。胱氨酸的溶解度随着尿pH值的增高而增加,因此,降低饮食中钠含量和动物蛋白的摄入可降低胱氨酸分泌[17,18]。推荐每日钠摄入量控制在2 g/d,动物蛋白摄入量控制在1 g/kg。增加口服液体量使尿量增加也是一个重要的治疗措施。治疗目标是将尿pH值控制在7.0以上使尿胱氨酸处于超饱和状态。本组3例患儿血液游离肉碱降低,精氨酸缺乏,存在继发性肉碱缺乏症及精氨酸缺乏[19]。左卡尼汀为重要的碱性维生素,可与体内蓄积的有机酸类物质结合,转化为水溶性的碱性的有机酸酯从尿中排泄,因此,对患儿补充了小剂量左卡尼汀。同时补充精氨酸,改善体质,

表 3 患儿及父母基因检测结果
并鼓励患儿多饮水,降低动物蛋白摄入量。为进一步碱化尿液,每日口服10%枸橼酸钾。经治疗后,3例患儿的病情均趋于稳定,均未发现肾结石增大或复发情况。尽管这些治疗措施可以降低胱氨酸分泌增加溶解度,但缺乏长期治疗效果的研究依据,需要进一步研究观察。
[1] Goodyer P, Boutros M, Rozen R. The molecular basis of cystinuria: an update [J]. Exp Nephrol, 2000, 8(3): 123-127.
[2] Scriver CR. Garrod's Croonian Lectures (1908) and the charter 'Inborn Errors of Metabolism': albinism, alkaptonuria,cystinuria, and pentosuria at age 100 in 2008 [J]. J Inherit Metab Dis, 2008, 31(5): 580-598.
[3] 贾建业, 陈方, 谢华, 等. 儿童胱氨酸结石的ESWL治疗[J]. 上海第二医科大学学报, 2005, 25(2)∶ 202-203.
[4] 邵建国, 侯飓, 宋飞, 等. 上尿路胱氨酸结石诊治22例报告[J]. 中国微创外科杂志, 2015, 15(7)∶ 622-623.
[5] Chace DH, Kalas TA, Naylor EW. The application of tandem mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism [J]. Annu Rev Genomics Hum Genet, 2002, 3: 17-45.
[6] Kimura M, Yamamoto T, Yamaguchi S. Automated metabolic profiling and interpretation of GC/MS data for organic acidemia screening: a personal computer-based system [J].Tohoku J Exp Med, 1999, 188(4): 317-334.
[7] Dello Strologo L, Pras E, Pontesilli C, et al. Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers:a need for a new classif i cation [J]. J Am Soc Nephrol, 2002,13(10): 2547-2553.
[8] Assimos DG, Leslie SW, Ng C, et al. The impact of cystinuria on renal function [J]. J Urol, 2002, 168(1): 27-30.
[9] Stephens AD. Cystinuria and its treatment: 25 years experience at St. Bartholomew's Hospital [J]. J Inherit Metab Dis, 1989, 12(2): 197-209.
[10] Lambert EH, Asplin JR, Herrell SD, et al. Analysis of 24-hour urine parameters as it relates to age of onset of cystine stone formation [J]. J Endourol, 2010, 24(7): 1179-1182.
[11] Harnevik L, Fjellstedt E, Molbaek A, et al. Mutation analysis of SLC7A9 in cystinuria patients in Sweden [J]. Genet Test,2003, 7(1): 13-20.
[12] Fattah H, Hambaroush Y, Goldfarb DS. Cystine nephrolithiasis [J]. Transl Androl Urol, 2014, 3(3): 228-233.
[13] Barbosa M, Lopes A, Mota C, et al. Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients [J]. Clin Genet, 2012, 81(1): 47-55.
[14] 谷现恩,闫治安,潘柏年,等. 胱氨酸结石16例诊治分析 [J]. 中华医学杂志 , 2003, 83 (15)∶ 1367.
[15] 谷现恩,梁丽莉. 尿石症的诊断与治疗[M]. 北京:人民卫生出版社,2008.
[16] Mattoo A, Goldfarb DS. Cystinuria [J]. Semin Nephrol, 2008,28(2): 181-191.
[17] Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria [J]. Kidney Int, 2006,69(6): 1041-1047.
[18] Jaeger P, Portmann L, Saunders A, et al. Anticystinuric effects of glutamine and of dietary sodium restriction [J]. N Engl J Med, 1986, 315(18): 1120-1123.
[19] Rasmussen J, Nielsen OW, Janzen N, et al. Carnitine levels in 26, 462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands [J]. J Inherit Metab Dis, 2014, 37 (2): 215-222.
The clinical and genetic findings of childhood cystinuria
MA Yanyan1, XIAO Haixue1, LIU Yupeng2, YUAN fumei1,LI Dongxiao2, SONG Jinqing2, LI Xiyuan2, DING Yuan2, YANG Yanling2(1. Qinghai University Affiliated Hospital, Xining 810001,Qinghai, China; 2. The First Hospital of Peking University, Beijing 100034, China)
Objective To explore the clinical features and genetic etiology of children with cystinuria with onset of kidney stone. Methods The clinical data of 3 children with cystinuria with onset of kidney stone and the gene analysis results of SLC3A1 and SLC7A9 by PCR sequencing were retrospectively analyzed. Results Three male children were from three unrelated families, kidney stone were presented in 2 cases at 1 year old and 1 case at 14 years old. The blood amino acid spectrum was normal in all 3 cases, while the free carnitine were decreased. The urinary amino acid spectrum indicated that cystine, ornithine,arginine, and threonine increased. Gene analysis conf i rmed that 1 case had homozygous mutations of SLC7A9 gene c.325G>A,and his parents were carriers of c.325G>A heterozygous mutation; other 2 cases had heterozygous mutations of SLC3A1 gene,c.1365delG and c.1113C>A heterozygous mutation in one case, and c.1897_1898insTA and c.1093C>T heterozygous mutation in one case, and their parents were heterozygous mutation carriers. After treatment with potassium citrate and L-carnitine, the conditions were improved in all cases. Conclusions Inherited metabolic disease should be considered for children with kidney stone. Urine amino acid analysis and gene detection are important methods for the diagnosis of cystinuria.
kidney stone; cystinuria; amino acid; inherited metabolic disease
doi∶10.3969/j.issn.1000-3606.2017.12.004
国家自然科学基金(No.81471097);国家自然科学青年基金(No.30872794);青海省应用基础研究项目(No.2016-ZJ-730)
杨艳玲 电子信箱:organic.acid@126.com
2017-04-05)
梁 华)
猜你喜欢
环境污染与防治(2021年11期)2021-01-09 02:08:51
临床儿科杂志(2020年2期)2020-03-12 04:04:10
发明与创新(2019年42期)2019-11-18 01:24:04
家庭医学(下半月)(2019年9期)2019-10-12 08:03:56
家庭百事通·健康一点通(2017年3期)2017-03-22 20:13:17
动物营养学报(2015年9期)2016-01-07 11:29:44
医学研究杂志(2015年3期)2015-06-10 06:41:52
中国医疗美容(2015年4期)2015-04-27 02:24:11
高中生学习·高一版(2014年6期)2014-07-05 10:20:16
中国优生优育(2014年8期)2014-01-20 08:26:45
