
熔融制样-X射线荧光测定锰矿中10种主次成分的条件优化
2017-12-28 01:00马景治
分析测试技术与仪器 2017年4期
马景治
(中南冶金地质研究所,湖北 宜昌 443003)
熔融制样-X射线荧光测定锰矿中10种主次成分的条件优化
马景治
(中南冶金地质研究所,湖北 宜昌 443003)
选用Li2B4O7、LiBO2和LiF(质量比为45∶10∶5)为混合熔剂,NH4NO3为氧化剂,LiBr为脱模剂,熔融制作样片,利用岛津1800型X 射线荧光光谱分析仪对GBW07261~GBW07266等锰矿标准物质拟合校准曲线,建立了 X 射线荧光同时测定锰矿中10种主次量组分(SiO2、Al2O3、Fe、MgO、CaO、K2O、Mn、TiO2、P、Zn)的快速分析方法. 对样品制备以及分析测试过程中的条件(熔剂稀释比例、熔融温度及时间、测定电压电流以及PHA等)进行了优化,在优化条件下,对GBW07263、GBW07265进行重复测定,相对标准偏差(RSD,n=12)低于2%. 同时对锰矿标准样品及合成锰矿样品进行分析,结果与参考值一致.
熔融制样;X射线荧光光谱法;锰矿;条件优化
锰矿石是冶炼锰和钢的重要工业原料,其各元素的含量直接影响到钢铁产品的质量,通常检测的项目有Mn、Fe、P、S、SiO2等[1-2]. 常规的分析手段主要有重量法、容量法、比色法等[3-5]. 这些分析方法的准确度高,重现性好,然而样品处理十分繁琐,并且分析结果容易受人为因素和试剂质量的影响[6].
X 射线荧光光谱法(XRF)作为一个成熟的分析技术广泛应用于冶金、水泥、石化和半导体等行业,其主次量组分的测定准确度可与化学分析方法媲美[7-11]. 本文采用1∶20熔剂比例稀释熔融处理样品,进一步消除了样品的粒度和成分的不均匀等影响,大大降低了基体的增强吸收效应和共存元素的干扰,拓宽了分析范围,适用于锰矿原材料的常规组分分析.
1 试验部分
1.1 仪器条件与试剂
XRF-1800(日本岛津):4.0 kW端窗铑靶X射线光管,最高工作电压60 kV,最大工作电流140 mA,75 μm铍窗;真空光路,扫描角度7°~148°,定位重现性0.000 1°.
Analymate-V4D高频感应熔样机(北京静远世纪科技有限责任公司);铂金坩埚(95% Pt+5% Au);分析天平(感重量0.1 mg).
Li2B4O7-LiBO2-LiF(质量比为45∶10∶5)优级纯混合熔剂(北京静远世纪科技有限责任公司),NH4NO3固体,LiBr(0.5 g/mL).
GBW07261~GBW07266(冶金部中南地勘局研究所);GBW07401(地矿部物化探所) ;H27~H29(冶金地质内部锰矿标准样品).
1.2 熔片制备
依次称取(6.000 0±0.000 4)g经过700 ℃灼烧2 h后的混合熔剂、(0.300 0±0.000 1)g经105 ℃烘2 h的锰矿标准样品和(0.300 0±0.010 0) g硝酸铵置于30 mL瓷坩埚中,搅拌均匀,转移至铂金坩埚中,加入0.5 mL 0.5 g/mL的LiBr溶液,用坩埚钳将坩埚放入熔样机内进行熔融制样. 熔融制样程序:在650 ℃下预热120 s,然后升温至950 ℃预熔240 s,前两阶段主要是将样品中的C、S除尽,保护坩埚,最后升温至1 050 ℃熔融180 s,冷却,脱模.
1.3 标准系列的配制
选用锰矿国家一级标准物质 GBW07261~GBW07266来建立标准校准曲线,为了提高基体组成的代表性并扩大某些元素的含量范围与分布,选用几个在基体组成、待测元素含量等方面有特点的标样与上述某些标样按不同比例相互混合,经充分混匀后,制得若干新的标准样品每个标准样品都具有一定的代表性,并构成一套具有一定梯度又有足够的含量范围的标准系列. 校准曲线含量范围如表1所列.

表1 校准样品各组分的含量范围Table1 Concentrationrangeofcomponentsincalibrationsamples
1.4 检测
根据标准曲线中各元素含量选择最高和最低的两个玻璃样片进行条件试验,选择最佳的电压、电流以及测试角度等条件,各元素的检测条件如表2所列.
2 结果与讨论
2.1 制样因素的优化
2.1.1 样品制备方式的选择
XRF分析粉末样品时常用的制样方法为压片法和熔片法. 压片法操作简单、经济,但是由于本体系标准样品复杂,存在严重的矿物效应,从而导致分析结果存在较大偏差[12]. 熔片法在操作上相对压片法较复杂,但在熔融的过程中,能够消除矿物效应和粒度效应,同时通过熔剂稀释,减小了基体效应,从而可以得到精密度和准确度都较好的分析结果[12]. 因此本文采用熔片法制样来测定锰矿石样品中的10种主、次量元素.
2.1.2 熔剂种类的选择
熔融制样时根据酸碱反应原理,熔剂应使用酸碱混合熔剂. 同时,应尽量满足熔成的玻璃片具有一定的机械强度、稳定和不易吸潮等特性. 本文采用Li2B4O7、LiBO2和LiF的混合试剂作为熔剂,其中LiF的加入使熔融流动性增强[13],从而使制备的样片更加均匀,同时通过机械转动使气泡在熔融阶段基本赶尽.
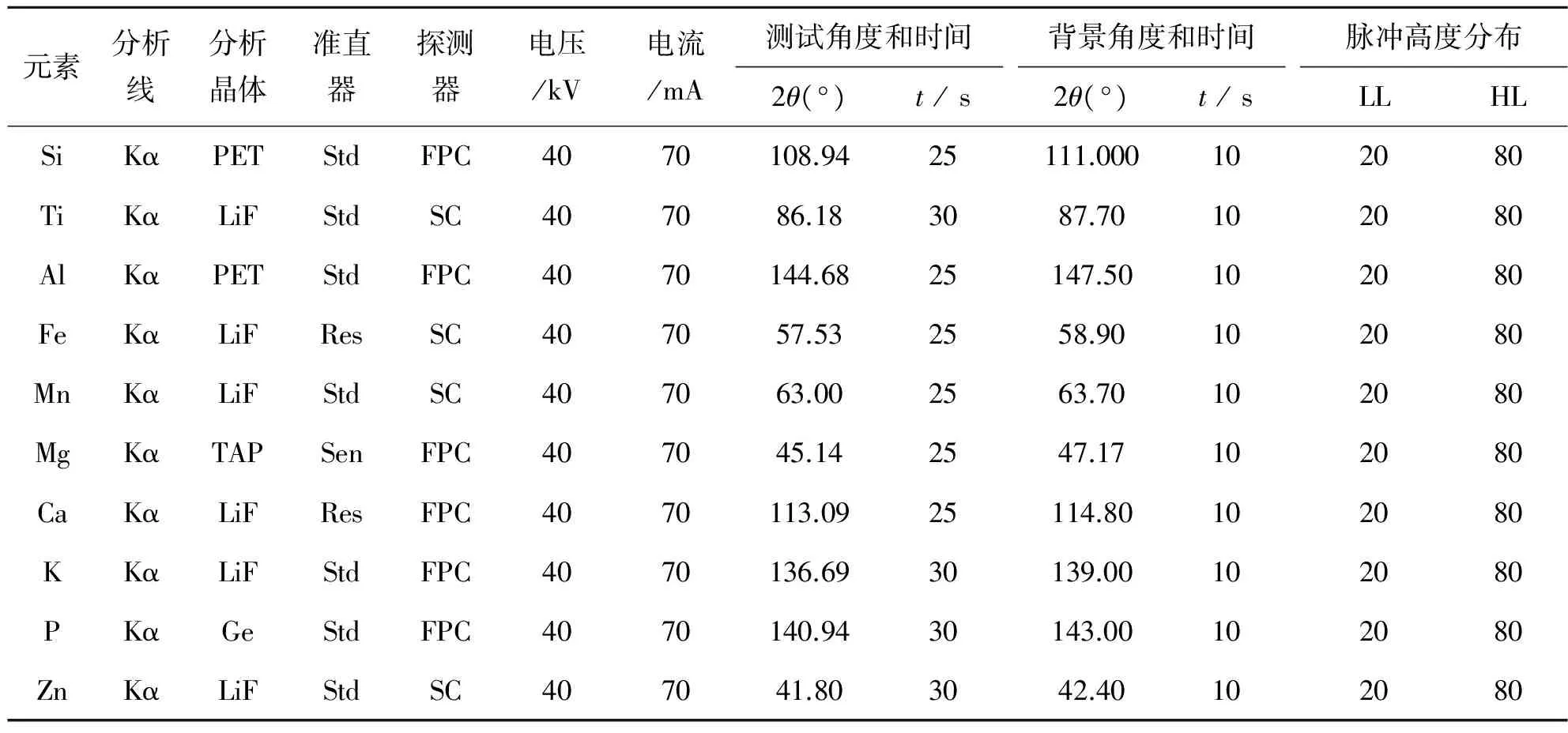
表2 各元素的最佳测量条件Table2 Optimalmeasuringconditionsofeachelement
2.1.3 熔剂稀释比的选择
样品和熔剂的配比直接影响样片的成型以及元素的荧光强度,分别采用Li2B4O7-LiBO2-LiF混合熔剂与样品的质量比为10∶1、15∶1、20∶1、25∶1、30∶1进行同一样品的制备,同时对样品进行XRF测定. 试验结果表明:当稀释倍数小于15时,流动性差,气泡较多,对坩埚的损害较大;当稀释倍数大于20时,玻璃片容易脱离坩埚,并随着稀释倍数的增加,元素的荧光信号强度逐渐降低,尤其对轻元素的测定不利. 试验选取GBW07265样品进行分析,图1示出了Si、Fe、Mn、Ca的信号强度随稀释比例的变化关系. 通过比较发现,在稀释比为20∶1时,能够得到相对稳定且具有一定信号强度的结果. 因此,本文采用20∶1的熔剂稀释比例.
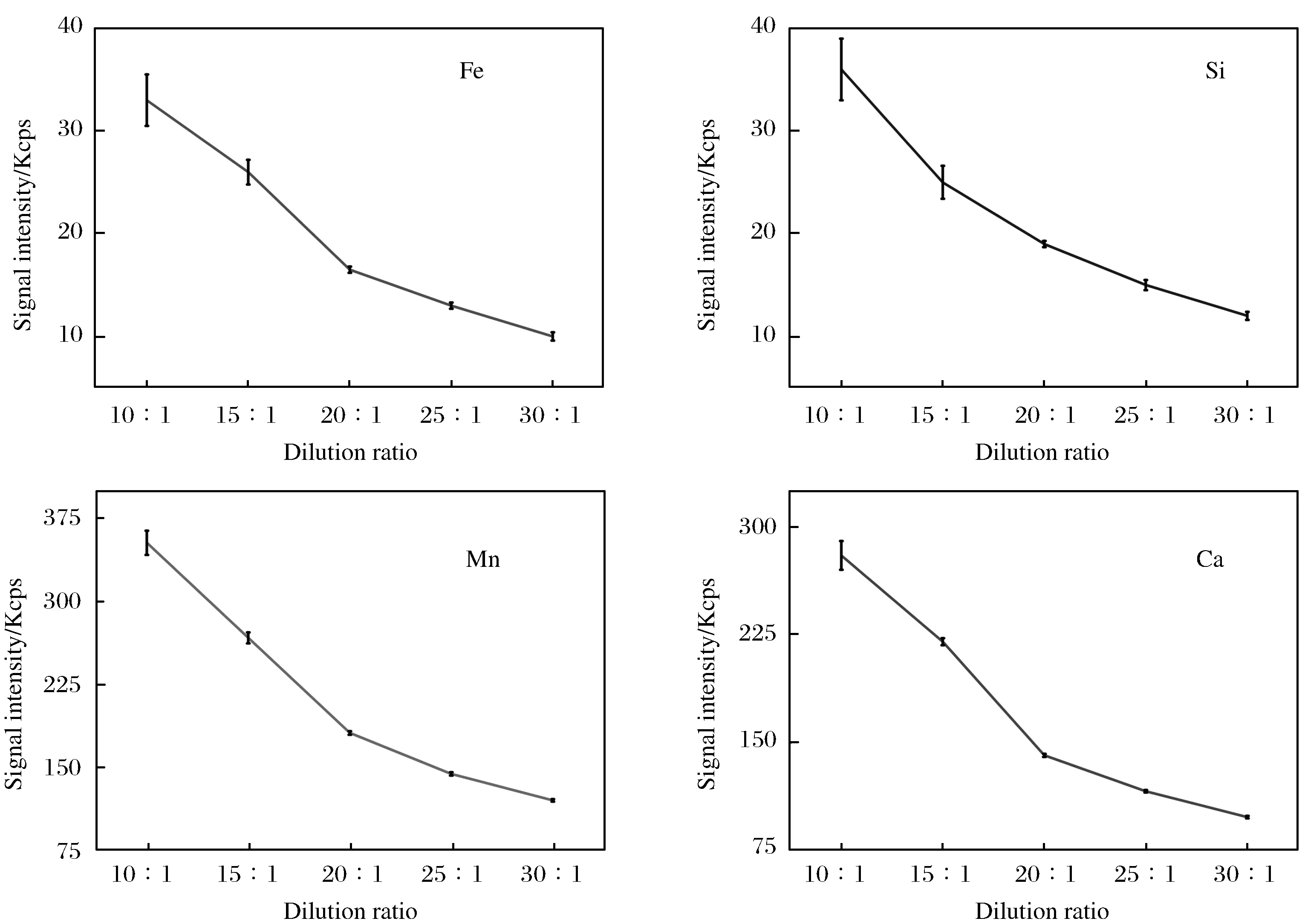
图1 稀释比例对信号强度的影响(n=3)Fig.1 Effect of dilution ratio on signal intensity (n=3)
2.1.4 脱模剂的选择及用量
常用的脱模剂有LiBr、NH4I、KBr和NaI等,在测定K、Na时我们不考虑KBr和NaI,而元素I对Ti的一次线存在干扰[12],因此本文采用LiBr为脱模剂.
大部分脱模剂在不同的熔融时间内可挥发,但仍会有残余,因此脱模剂的加入量既要保证熔融过程具有好的流动性和脱模性,又要尽量少的加入脱模剂. 试验分别加入0.1、0.3、0.5、0.7、0.9 mL LiBr脱模剂(0.5 g/mL)对GBW07263进行多次熔样测定,图2示出了Si、Fe、Mn、Ca的信号强度随脱模剂加入量的变化情况. 为了得到稳定性较好的结果,最终选用0.5 mL 0.5 g/mL的LiBr作为脱模剂的加入量.

图2 脱模剂加入量对信号强度的影响(n=3)Fig.2 Effect of quantity of mold release agent on signal intensity (n=3)
2.1.5 熔融温度的选择
对同一样品分别在950、1 000、1 050和 1 100 ℃下进行熔融. 通过试验发现,当熔融温度低于1 000 ℃时,熔融制得的样品不够均匀,在熔融过程中熔液流动性差;在1 050和1 100 ℃下制备的样片光滑,熔片不易裂. 考虑到高温对坩埚的影响,本试验选择1 050 ℃作为熔融温度.
2.2 方法的检出限
各分析组分的检出限LLD(仪器检出限)按照式(1)计算,计算结果如表3所列.
式中,m:测量灵敏度(标准曲线斜率);Ib:背景的荧光强度;Tb:背景的测量时间.
方法检出限与样品中基体含量有关,不同的样品因组分含量不同,背景强度也会不同,因此检出限也不同. 本文采用同一个锰矿样品进行熔片,连续测定12次,求出各组分的标准偏差,以3倍标准偏差作为本法的检出限,结果如表3所列.
2.3 基体效应与谱线重叠校正
通过大比例熔剂稀释熔融制样在一定程度上可降低基体的增强吸收效应,然而由于样品中主次成分含量差异大,范围较宽,有些元素之间的增强-吸收效应难以消除,因此必须进行基体校正. 本试验采用仪器自带的基体校正公式进行计算相应的吸收-增强系数以及谱线重叠系数,计算公式如式(2):
Wi=a×I2+b×I+c×1+∑dj×Wj-∑Lj×Wj(j≠i)
(2)
式中:Wj—基体元素定量结果;dj—吸收影响系数;Lj—重叠影响系数;Wi—被校正元素的定量结果;I—被校正元素的X射线强度;a,b,c—标准曲线常数.

表3 各组分的检出限Table3 Detectionlimitsandquantificationlimitsofcomponents /(μg/g)
2.4 精密度分析
按照1.2节制样方法,分别将锰矿标准样品GBW07263和GBW07265重复制备12个样片,按照表2的测量条件进行分析,统计结果列于表4.
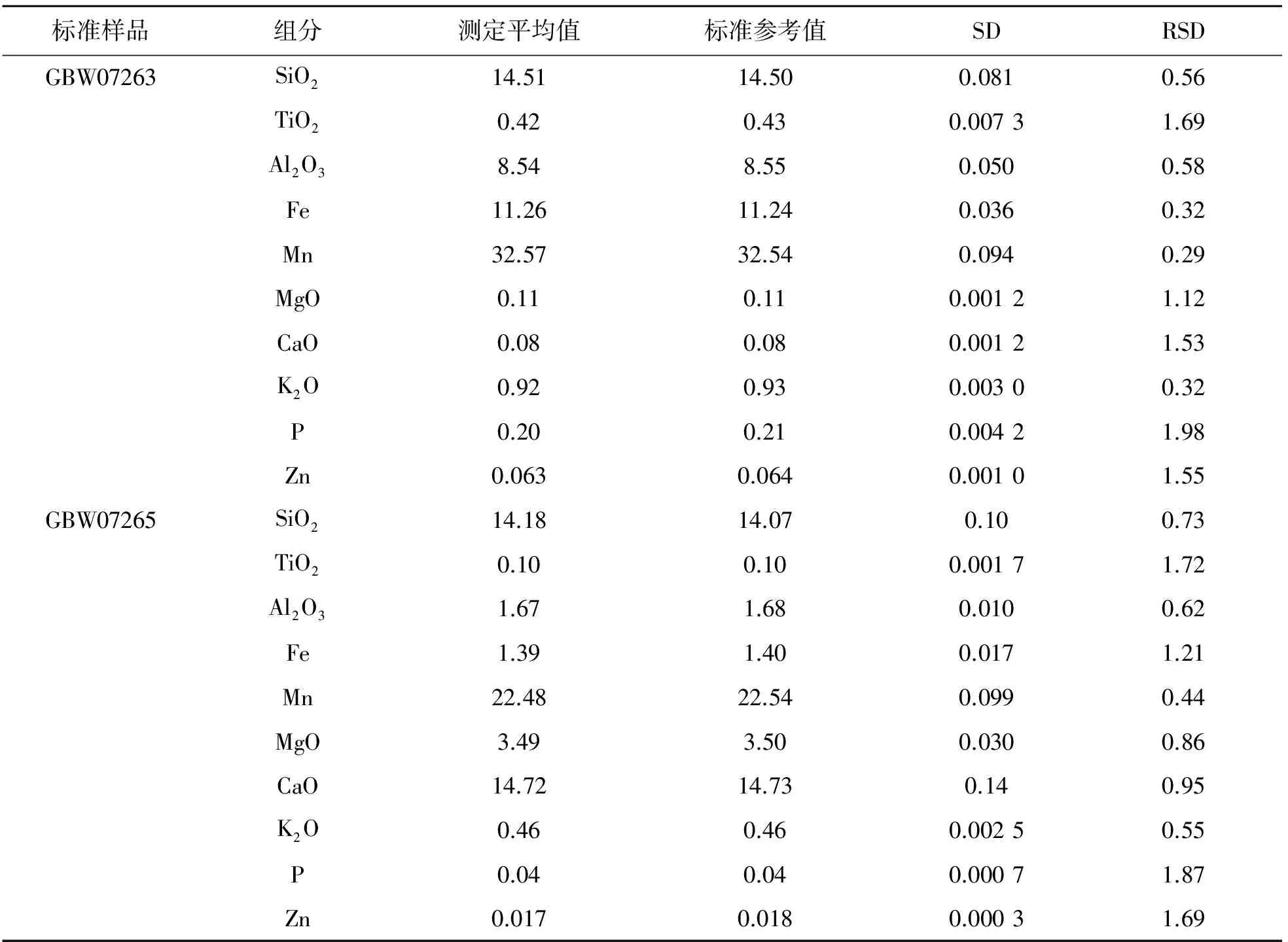
表4 精密度试验(n=12) Table4 Precisiontest(n=12) /%
2.5 准确度分析
为了验证该方法的准确性以及实用性,分别对锰矿标准样品GBW07263、GBW07264、GBW07265以及混合标准样品进行测试,分析结果如表5所列. 从表5可以看出,采用本文选定的试验条件进行熔融制样,所得样片通过X射线荧光光谱仪分析,所得结果与标准值有良好的一致性,表明该方法检测结果准确可靠.
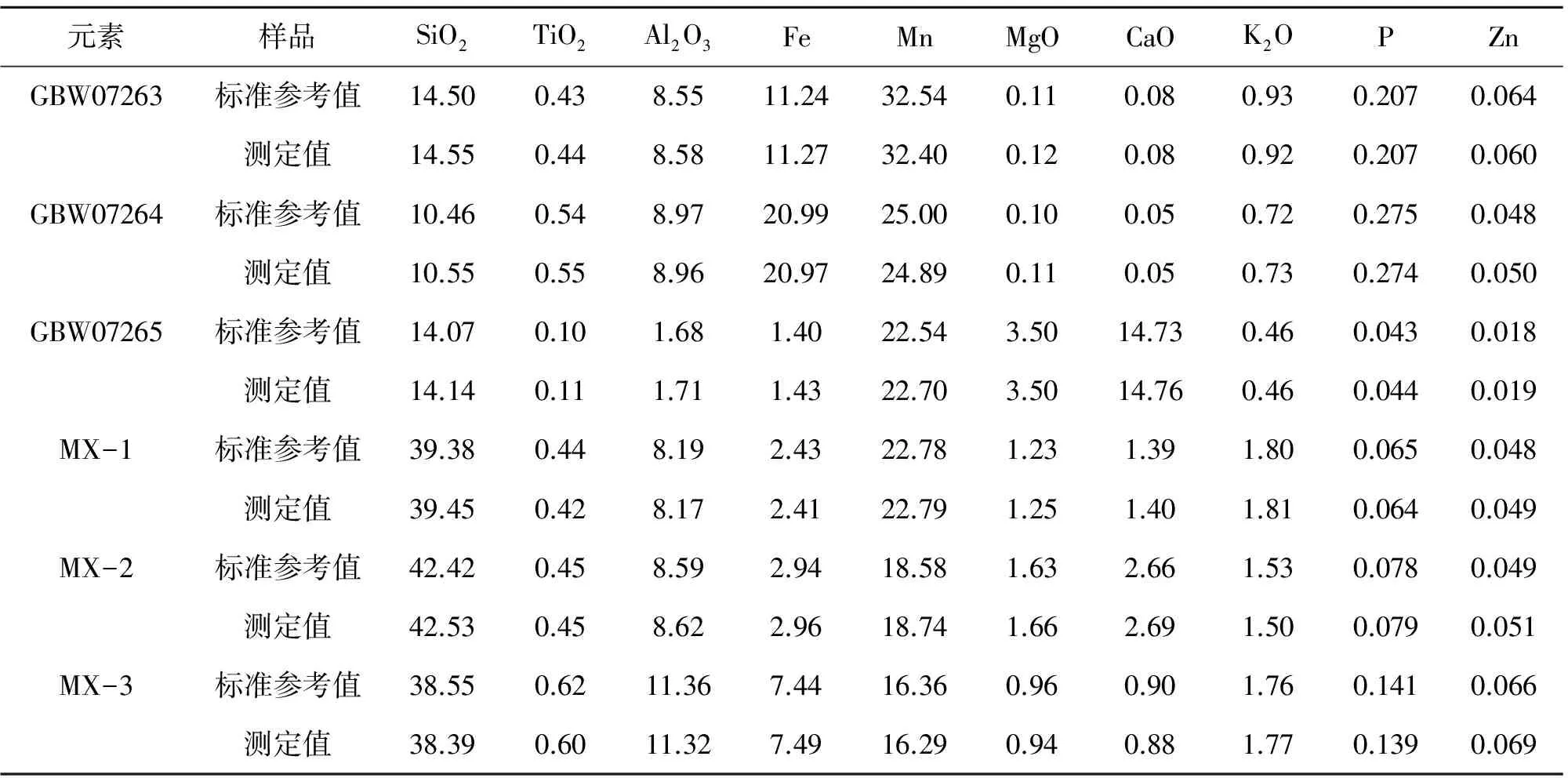
表5 准确度试验(n=3) Table5 Accuracytest (n=3) /%
备注:MX-1(GBW07261∶GBW07401=1∶1);MX-2(GBW07262∶GBW07401=1∶1);MX-3(GBW07263∶GBW07401=1∶1)
3 结论
本文采用Li2B4O7、LiBO2和LiF的混合熔剂进行熔融制样,建立了XRF同时测定锰矿样品中10种主次量组分的方法. 该方法适用的含量范围宽、准确度高,与常规的化学分析相比,操作简便快速、绿色环保,适合实际样品的批量测定.
[1] 岩石矿物分析编写组. 岩石矿物分析(第二分册)[M]. 北京: 地质出版社, 2011: 804-861.
[2] 朱明娟. 锰矿成分的分析方法及应用实践微探[J]. 中国锰业, 2017, 35(1): 116-118.
[3] 周伟,张楠,赵祝鹏,等. 高碘酸钾氧化光度法测定含大量铁(Ⅱ)的锰矿浸出液中锰[J]. 冶金分析, 2013, 33(2): 47-50.
[4] 张悫,邓军华,王一凌,等. 容量法快速测定锰矿中二氧化硅含量[J]. 中国锰业, 2012, 30(1): 41-43.
[5] 焦立为,李海涛. ICP-AES测定锰矿中二氧化硅含量[J]. 光谱实验室, 2007, 24(3): 360-362.
[6] 唐梦奇,黎香荣,魏亚娟,等. X射线荧光光谱法测定烧结锰矿中的主次量成分[J]. 光谱实验室, 2012, 29(2): 977-981.
[7] 刘江斌,党亮,和振云. 熔融制样—X射线荧光光谱法测定锰矿石中17种主次组分[J]. 冶金分析, 2013, 33(9): 37-41.
[8] 刘江斌,祝建国. X射线荧光光谱法快速测定锰矿石中的主次组分[J]. 分析测试技术与仪器, 2012, 18(1): 34-37.
[9] 曲月华,王一凌,张悫,等. 熔融制样-X射线荧光光谱法测定锰矿中9种组分[J]. 冶金分析, 2011, 31(9): 24-29.
[10] 王谦,林力,朱丽辉,等. 应用理论α系数校正X射线荧光光谱法分析锰矿时的基体效应[J]. 理化检验(化学分册), 2008, 44(7): 658-660.
[11] 李小莉. 熔融制片-X射线荧光光谱法测定锰矿样品中主次量元素[J]. 岩矿测试, 2007, 26(3): 238-240.
[12] 马景治,刘恒杰,王峰. 熔融制样-X射线荧光光谱法测定地质样品中的主次成分[J]. 分析试验室, 2016, 35(11): 1348-1352.
[13] 马景治,贾海峰,兰绿灯,等. X射线荧光光谱法测定硅石中主次量元素组分[J]. 中国无机分析化学, 2017, 7(2): 55-58.
ConditionOptimizationofX-rayFluorescenceSpectrometrywithFusionSamplePreparationforDeterminationofTenMajorandMinorComponentsinManganeseOres
MA Jing-zhi
(CentralSouthInstituteofMetallurgicalGeology,Yichang443003,HubeiChina)
A fast and simple approach to directly determine SiO2、Al2O3、Fe、MgO、CaO、K2O、Mn、TiO2、P and Zn in manganese ores samples by X-ray fluorescence spectrometer(XRF)was developed. This method used a glass fuse piece technique with Li2B4O7、LiBO2and LiF (Mass ratio:45∶10∶5) as the flux, NH4NO3as the oxidant, LiBr as the stripping agent. Several experimental parameters were optimized, and the analytical figures of merit were evaluated. The components in the certified reference material of GBW07261~GBW07266 were determined by the proposed method. The relative standard deviation (RSD,n=12) was less than 2%. The accuracy test was conducted with synthetic samples. The results were consistent with the certified values.
fusion sample preparation; XRF; manganese ores; condition optimization
分析测试新成果(261~266)
2017-08-10;
2017-09-07.
马景治(1990-),男,硕士研究生,助理工程师,主要从事岩石矿物无机元素分析,E-mail:cugedu@163.com.
O657.32
B
1006-3757(2017)04-0261-06
10.16495/j.1006-3757.2017.04.009
猜你喜欢
山东冶金(2022年2期)2022-08-08
轮胎工业(2022年3期)2022-07-20
广东建材(2021年8期)2021-08-13
陶瓷学报(2021年3期)2021-07-22
矿山安全信息(2021年25期)2021-01-03
应用技术学报(2020年2期)2020-02-21
化学分析计量(2016年1期)2016-03-14
铜业工程(2015年4期)2015-12-29
黑龙江交通科技(2015年4期)2015-03-21
化工生产与技术(2014年3期)2014-02-27
