
草地早熟禾干旱胁迫转录组差异性分析
2017-12-22 01:52冷暖刘晓巍张娜许立新
草业学报 2017年12期
冷暖,刘晓巍,张娜,许立新*
(1.北京林业大学草坪研究所, 北京 100083;2.南京市老山林场, 江苏 南京 211811)
草地早熟禾干旱胁迫转录组差异性分析
冷暖1,刘晓巍2,张娜1,许立新1*
(1.北京林业大学草坪研究所, 北京 100083;2.南京市老山林场, 江苏 南京 211811)
干旱是影响草地早熟禾生产力的主要因素之一。在没有参考基因组的情况下, 为了揭示草地早熟禾在干旱处理下基因表达谱的变化, 本文采用了高通量Illumina Hiseq测序平台对草地早熟禾的对照组(CK)与干旱处理组(D)进行转录组测序, 并对测序数据进行了分析研究, 进一步探究了草地早熟禾干旱应答的分子机制。结果表明, 在干旱处理下共检测到24465个差异表达的基因, 筛选后获得4143个上调基因和4415个下调基因, 共占差异表达基因总数的34.98%。经富集分析后得, 与蛋白激酶、蛋白磷酸酶、碳代谢以及ABA等相关的基因可以作为研究草地早熟禾干旱响应机制的主要研究对象。qRT-PCR分析表明, 随机选出的8个差异性表达基因的表达趋势与高通量测序结果相一致。此外, 还候选了吲哚-3-甘油磷酸合成酶、蛋白磷酸酶、已糖激酶、钙结合蛋白、叶绿素a/b结合蛋白等基因作为与草地早熟禾干旱胁迫相关的应答候选基因, 为揭示草地早熟禾耐旱分子机制奠定了基础。
草地早熟禾;干旱胁迫;高通量测序;转录组
在我国北方, 草地早熟禾是一种适应性很强的冷季型草, 品质好, 繁殖快, 广泛应用于公园、居住区、运动场等地绿化。但我国北方大部分地区为干旱半干旱地区, 不同地区面临着不同程度的缺水问题, 所以干旱成为影响草地早熟禾在北方生长发育的主要因素之一。水分是植物体的重要组成部分, 它参与植物的光合、呼吸、蒸腾等生理作用, 也是影响植物形态结构等重要生物因子, 充足的水分供应是植物正常生存的重要条件。干旱胁迫时, 植物为了适应环境, 不仅在生理上会发生反应, 分子上也会产生一定的变化, 一些功能不同的基因在干旱胁迫时表达量会发生上调或者下调。李伟[1]发现, 草地早熟禾(Poapratensis)PpNAC与水稻(Oryzasativa)的NAC蛋白SNACI高度同源, 水稻的SNACI可使植株抗旱性提高, 因而推测PpNAC与草地早熟禾的抗旱性有密切联系;Xu[2]发现, 干旱会诱导草地早熟禾两个品种‘Midnight’和‘Brilliant’的Cyt Cu/ZnSOD、Chl Cu/ZnSOD和APX三个基因上调表达, MR基因下调表达, 且与‘Brilliant’相比, 抗旱性好的‘Midnight’品种这4个基因差异表达更显著;信金娜[3]通过基因枪轰击法把抗旱、耐盐有关外源基因(DREB1A、BADH-CMO、CMO)转入草地早熟禾, 获得转基因植株, 结果表明, 转基因植株在抗旱性方面强弱程度为转双基因植株>转DREB1A、CMO基因植株>对照。抗旱性是一个多基因控制的数量性状[4], 因而有很多优良基因还没有能够完全被准确探明。
Illumina测序属于二代测序技术, 它是基于特定限制酶切位点的测序分析, 不需要知道基因组序列, 适于研究基因组不明确的物种的基因表达[5], 并且具有测序通量大、成本低的优势。目前, 在黑麦草(Loliumspp.)[6]、紫花苜蓿(Medicagosativa)[7]、结缕草(Zoysiajaponica)[8-9]以及草地早熟禾[10]的研究中都有应用, 通过测序技术获得草坪草不同胁迫环境的响应基因并进行基因表达差异性分析。因此, Illumina测序技术是研究草地早熟禾干旱胁迫基因差异表达较好的选择。
本研究中运用Illumina高通量测序平台的转录组测序技术, 分别对正常水分条件和干旱条件下生长的草地早熟禾成熟叶片的转录产物mRNA进行测序,测得的数据能够较全面反映草地早熟禾干旱胁迫下基因表达状况, 分别对差异表达基因进行GO、KEGG(Kyoto Encyclopedia of Genes and Genomes)注释和富集分析, 进一步筛选出与草地早熟禾抗旱相关的基因, 为之后草地早熟禾SSR标记开发、抗旱性相关基因的发掘及分子育种提供借鉴。
1 材料与方法
1.1 实验材料与处理
以草地早熟禾的‘Nuglade’品种为材料, 购自北京绿冠公司。草坪养护在北京林业大学八家村实验站温室中进行。试验于2016年1月开始,设置两种处理, 对照处理和干旱胁迫处理。对照处理的植株2 d浇一次水, 保证土壤含水量在35%;干旱胁迫处理组不浇水, 直至土壤水分降到4%。每个处理3次生物学重复。以正常浇水的材料为对照样本(CK), 干旱胁迫为处理样本(D)。在干旱胁迫后第15天的8:30-9:00之间对对照组(CK)和干旱处理组(D)进行取样。
1.2 文库的构建及库检
按照TRIzol植物RNA提取试剂盒方法分别提取草地早熟禾对照组(CK)与干旱处理组(D)样品叶片的RNA。所提取的RNA经电泳检测合格后送至北京诺禾致源生物有限公司进一步确认质量, 合格后利用 Oligo(dT)磁珠富集真核生物mRNA, 并以 Oligo (dT)引导反转录合成双链 cDNA, 通过PCR扩增和AMPure XP beads纯化后获得最终文库。文库构建完成后, 使用Qubit2.0进行初步定量, 用Agilent 2100对文库进行检测, 符合预期后, 使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2 nmol/L), 以保证文库质量。库检合格后, 进行Illumina HiSeq测序。
1.3 Illumina测序及测序数据的分析
1.3.1测序原始数据质量评估与转录本拼接 高通量测序(如Illumina HiSeqTM)得到的原始图像数据文件经CASAVA碱基识别(base calling)分析转化为原始测序序列(sequenced reads), 称之为 Raw Data或Raw Reads。通过测序错误率分布检查, A/T/G/C含量分布检查, 进行c测序数据过滤后得到Clean Reads。对于无参考基因组的项目, 获得Clean Reads后, 需要对Clean Reads进行拼接以获取后续分析的参考序列。采用Trinity对Clean Reads进行拼接[11]。将Trinity拼接得到的转录本序列, 作为后续分析的参考序列。取每条基因中最长的转录本作为Unigene, 以此进行后续的分析。
1.3.2基因功能注释 为获得全面的基因功能信息, 进行了七大数据库的基因功能注释, 包括: Nr, Nt, PFAM, KOG/COG, Swiss Prot, KEGG, GO。得出数据在七大数据库中的注释成功率情况。
1.3.3差异表达基因分析 统计好样本中基因表达量后, 分别对对照组(CK)与干旱处理组(D)之间基因表达情况进行对比, 找出表达量存在差异的基因, 并通过筛选进一步发掘出显著差异表达的基因。对于有生物学重复的样品, 采用DESeq[12]进行分析, 筛选阈值为p-adjusted<0.05。
1.3.4差异表达基因富集分析 GO功能显著性富集分析给出与基因组背景相比, 在差异表达基因中显著富集的GO功能条目, 从而给出差异表达基因与哪些生物学功能显著相关。GO富集分析方法为 GOseq[13],此方法基于Wallenius non-central hyper-geometric distribution。在生物体内, 不同基因相互协调行使其生物学功能, 通过Pathway显著性富集能确定差异表达基因参与的最主要生化代谢途径和信号转导途径。KEGG是有关Pathway的主要公共数据库[14]。
1.4 qRT-PCR转录组数据验证
随机选取8个差异性表达的基因进行qRT-PCR分析, 对高通量数据进行验证。分别提取对照组(CK)与干旱组(D)的叶片RNA, 采用HiScript® Ⅱ Q RT SuperMix for qPCR试剂盒反转录合成cDNA, 设计引物(表1)进行qRT-PCR实验, Actin基因作为内参。各个处理均做3次重复, 计算基因的相对表达量。

表1 干旱胁迫条件下草地早熟禾8个差异性表达基因qRT-PCR引物Table 1 Primers of eight differentially expressed genes used for quantitative real-time PCR in P. pratensis at drought stress
2 结果与分析
2.1 测序数据统计与评估
测序得到的原始测序序列Raw Reads中含有带接头的、低质量的Reads, 对照及各处理组的Raw Reads都在4052万条以上。在去除带接头的Reads、去除N的比例大于0.1%的Reads、去除低质量Reads (质量值Qphred≤20的碱基数占整个Reads的50%以上的Reads) 后, 得到Clean Reads, 后续分析都基于Clean Reads。经过滤后, 共得到59.35G的有效Reads (Clean Reads)。由表2可知, 各组得到的Clean Reads的量占原始Reads的比例都达到了96%以上, 说明测序的建库工作质量良好。
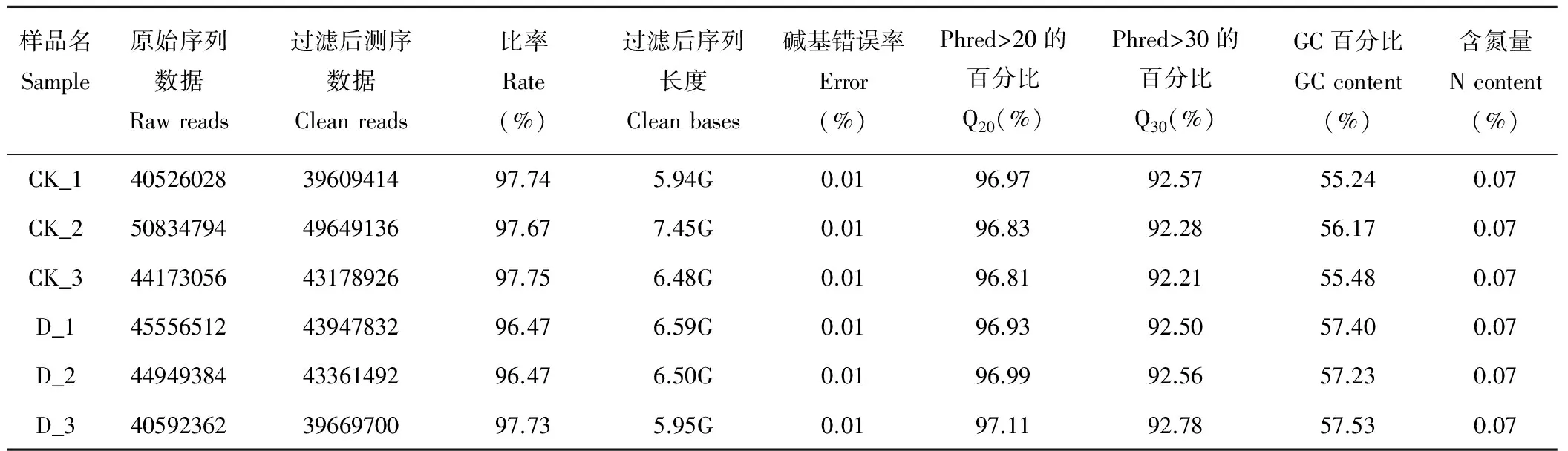
表2 测序数据输出质量情况Table 2 Sequencing data quality evaluation
2.2 转录组的拼接
拼接所得的序列长度用于衡量拼接质量, 如拼接所得的序列长度越长, 则测序质量越好。经Trinity拼接后, 获得转录本(Transcript)435250条, 总长度为324271434 nt,平均长度为745 nt, N50长度为1163 bp;Unigene 254331条, 总长度为147657025 nt,平均长度为581 nt, N50长度为818 bp。具体的组装统计结果见表3。由表3数据可知, 拼接所得片段有很高的组装完整性, 拼接所得序列长, 测序质量好。
2.3 基因功能注释
为了预测Unigene的功能, 对其进行基因注释, 具体方法为将Unigene序列分别与主要的生物学数据库, 如GO、KEGG、NR等进行基因信息比对, 最终有254331条Unigene获得注释, 其中, NR数据库注释的信息最多, 占全部Unigene的30.58%。7842条Unigene同时比对到7个数据库中, 占总数的3.08%。105852条Unigene至少在一个数据库中得到功能注释, 占到总数的41.61%。结果如图1所示。

表3 Unigene与转录本的长度统计结果表Table 3 Unigene and transcript length statistics
N50: 将组装片段按从长到短排序, 并进行长度值累加, 当长度值累计至总长度的50%时, 最后累加的片段长度值。Unigene: 取每条基因中最长的转录本作为该基因的代表, 称为Unigene, 以此进行后续的分析。nt: 核苷酸单位。N50: The assembled segments are sorted from long to short and the length values are accumulated. When the length value is accumulated to 50% of the total length. The last accumulated fragment length value. Unigene: The longest transcript of each gene was used as a representative of the gene, called Unigene, followed by subsequent analysis. nt: The unit of nucleotides.
2.4 基因差异性表达分析

图1 Unigene在各大数据库中的功能注释分布柱状图Fig.1 Distribution histogram of Unigene functional annotation according to the major databases
对对照组(CK)与干旱组(D)进行差异基因筛选(DEGs), 经筛选, 共鉴定出24465个差异表达的基因, 其中上调基因有12783个, 占差异表达基因总数的52.25%;下调基因共11682个, 占差异表达基因总数的47.75%。结果如图2所示。
2.5 差异表达基因的GO分析
GO 分析(gene ontology)是一个常用的基因功能分类体系, 它按照生物途径(biology process), 分子功能(molecular function)和细胞组分(cellular component)对基因进行注释和分类。通过对差异表达基因进行GO terms富集度统计学的分析, 计算出差异基因GO term的p-value和p-value的FDR值(q-value), 定位差异基因最可能相关的GO term。GO 分析对实验结果有提示的作用, 通过差异基因的GO 分析, 可以找到富集差异基因的GO分类条目, 寻找不同样品的差异基因可能和哪些基因功能的改变有关。
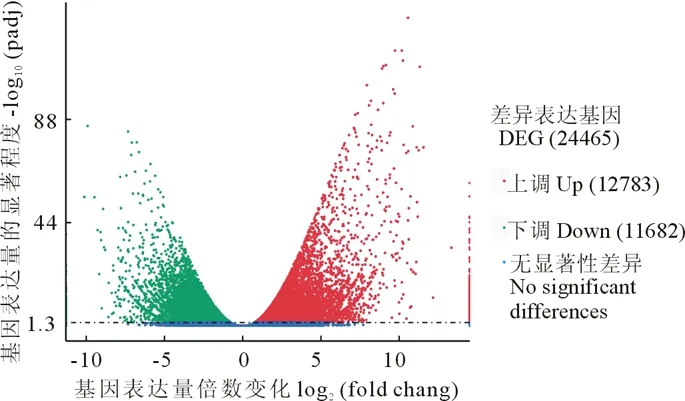
图2 干旱处理(D)/对照组(CK)基因差异表达分析火山图Fig.2 Volcanic map of differential gene expression analysis between drought treatment (D)/control group (CK)
对干旱组(D)与对照组(CK)之间的差异表达基因(DEGs)进行GO功能分析得到图3, 经分析, 代谢过程(metabolic process)、蛋白磷酸化(protein phosphorylation)、泛素连接酶复合物(ubiquitin ligase complex)、催化活性(catalytic activity)和蛋白激酶活性(protein kinase activity)等与干旱胁迫紧密相关的生物学途径、分子功能、细胞组成等都有明显的响应变化。

图3 差异表达基因GO富集分类Fig.3 GO enrichment classification of differentially expressed genes 1: 响应水 Response to water;2: 大分子修饰 Macromolecule modification;3: 蛋白磷酸化 Protein phosphorylation;4: 细胞蛋白修饰过程 Cellular protein modification proess;5: 蛋白修饰过程 Protein modification process;6: 响应非生物刺激 Response to abiotic stimulus;7: 响应无机物质 Response to inorganic substance;8: 蛋白代谢过程 Protein metabolic process;9: 响应含氧化合物 Response to oxygen-containing compound;10: 磷酸化 Phosphorylation;11: 响应酸性化学品 Response to acid chemical;12: 代谢过程 Metabolic process;13: 细胞蛋白质代谢过程 Cellular protein metabolic process;14: 磷酸代谢过程 Phosphorus metabolic process;15: 含磷酸盐的化合物代谢 Phosphate-containing compound metabolic;16: 蛋白泛素化 Protein ubiquitination;17: 小蛋白质共轭修饰 Protein modification by small protein conjugation;18: 小蛋白质共轭或去除修饰 Protein modification by small protein conjugation or removal;19: 过期过氧化酶反应 Obsolete peroxidase reaction;20: 脂质生物合成过程 Lipid biosynthesis process;21: 泛素连接酶复合物 Ubiquitin ligase complex;22: Cullin-RING泛素连接酶复合物 Cullin-RING ubiquitin ligase complex;23: 核泛素连接酶复合物 Nuclear ubiquitin ligase complex;24: 后期促进复合物 Anaphase-promoting complex;25: 脂肪酸合酶复合物 Fatty acid synthase complex;26: 光系统Ⅱ氧气演变复合体 Photosystem Ⅱ oxygen evolving complex;27: 转移酶活性 Transferase activity;28: 催化活性 Catalytic activity;29: 蛋白激酶活性 Protein kinase activity;30: 磷酸转移酶活性, 酒精组作为受体 Phosphotransferase activity, alcohol group as acceptor;31: 激酶活性 Kinase activity;32: 泛素蛋白转移酶活性 Ubiquitin-protein transferase activity;33: 泛素类似蛋白转移酶活性 Ubiquitin-like protein transferase activity;34: 血红素结合 Heme binding;35: 四吡咯结合 Tetrapyrrole binding;36: 转移酶活性, 转移含磷基因 Transferase activity, transferring phosphorus-containing groups;37: ATP结合 ATP binding;38: 转移酶活性, 转移除氨基酰基以外的酰基 Transferase activity, transferring acyl groups other than amino-acyl groups;39: 嘌呤核糖核苷三磷酸结合 Purine ribonucleoside triphosphate binding;40: 脂肪酸合酶活性 Fatty acid synthase activity;41: 转移酶活性, 转移己糖基 Transferase activity, transferring hexosyl groups;42: 离子结合 Ion binding;43: 3-氧代酰基-[酰基-载体-蛋白]合酶活性 3-oxoacyl-[acyl-carrier-protein] synthase activity;44: 腺苷酸核糖核酸结合 Adenyl ribonucleotide binding;45: 过氧化物酶活性 Peroxidase activity;46: 腺苷酸核苷酸结合 Adenyl nucleotide binding.
其中, 4143个上调Unigenes, 有2416个(58%)Unigenes注释到生物学过程(BP), 其中富集较显著的分布在蛋白质改性过程(protein modification process)与单生物体运输(single-organism transport);543个(13%)Unigenes注释到细胞组分(CC), 其中富集较显著的为nucleus;1181个(29%)Unigenes注释到分子功能(MF), 其中富集较显著的为转移酶活性(transferase activity)和阳离子活性(cation activity)。
4415个下调Unigene, 有2643个(60%)Unigenes注释到生物过程(BP), 其中富集最显著的为代谢过程(metabolic process)和大分子代谢过程(macromolecule metabolic process);550个(12%)Unigenes注释到细胞组分(CC), 1217个(28%)Unigenes注释到分子功能(MF), 均匀分布在20类基因功能中。
2.6 差异表达基因KEGG富集分析
KEGG是系统分析基因功能、基因组信息的数据库, 可用于进行生物体内代谢分析与代谢网络研究。为对样本进行全面的代谢通路分析, 将测序所得Unigene序列比对到KEGG数据库。不同数据库注释基因数目的统计结果显示, 共有20398条Unigenes在KEGG数据库中得到注释, 占总数的8.02%, 参与到129个代谢途径中, 其中注释到植物-病原体相互作用(plant-pathogen interaction)中的最多, 为848个。
对样本对照组(CK)与干旱组(D)之间的差异表达基因(DEGs)进行KEGG代谢通路分析后, 共获得差异表达基因(DEGs)8764个, 对应在307个代谢通路中。
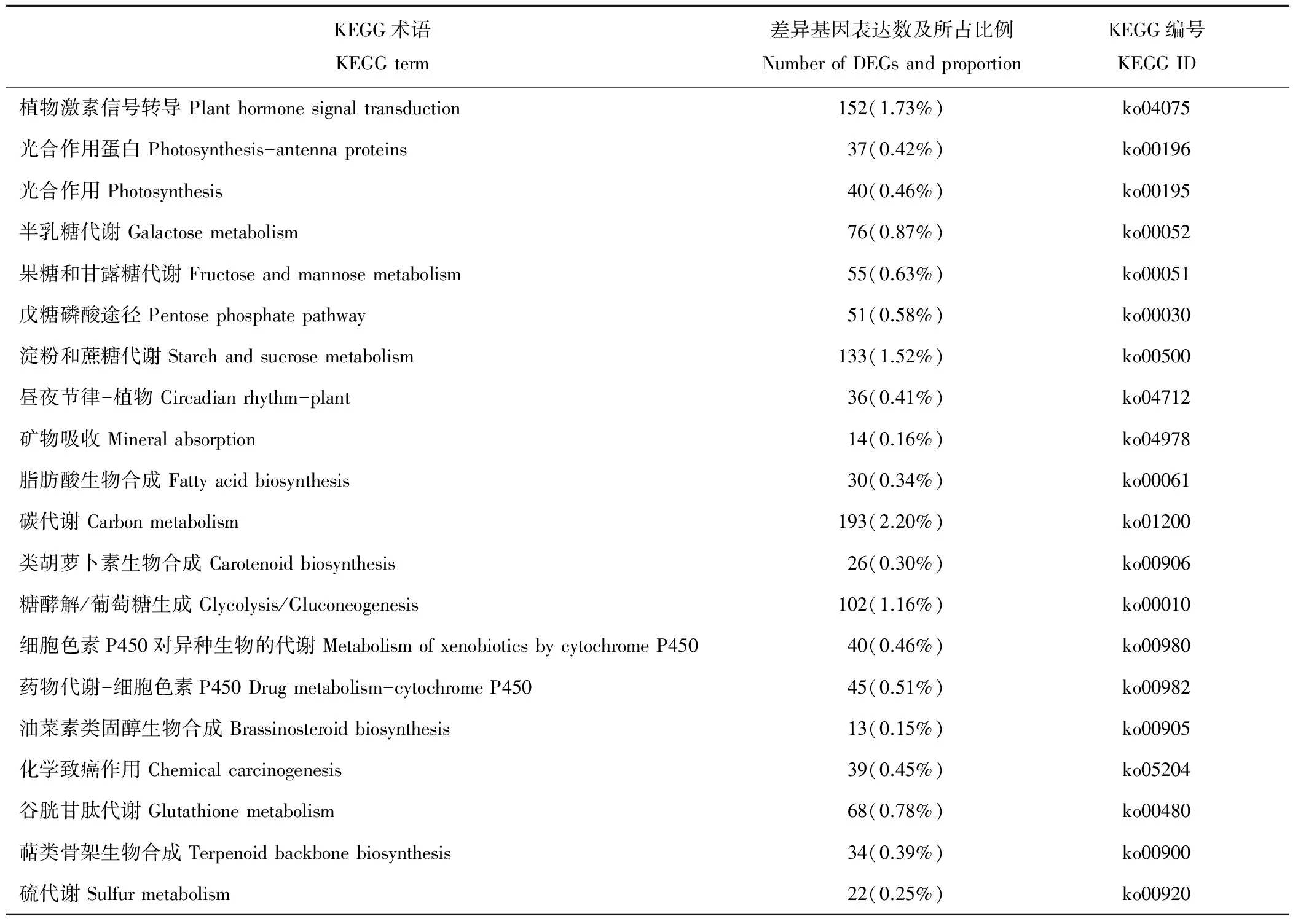
表4 草地早熟禾干旱胁迫下差异基因富集程度排名前20的Pathway条目Table 4 Top 20 enrichment pathway in responses to drought in Poa pratensis

图4 差异表达基因KEGG富集分析Fig.4 KEGG enrichment analysis of differentially expressed genes
进一步统计后发现, 在307个KEGG Pathway中最显著的4个Pathway的是植物激素信号转导(plant hormone signal transduction)、光合作用蛋白(photosynthesis-antenna proteins)、光合作用(photosynthesis)以及半乳糖代谢(galactose metabolism)。 由表4和图4可知, 排名前20显著性富集差异基因占差异表达基因总量的13.77%, 主要涉及代谢途径、信号转导以及次生代谢等。其中, 与碳代谢相关的基因占差异表达基因总量的2.20%, 与植物激素信号转导相关的基因占1.73%, 淀粉和蔗糖代谢相关的基因占1.52%, 糖酵解相关的基因占1.16%, 其他相关基因所占比例相对较低。差异性表达基因在植物激素信号转导和碳代谢的通道上富集程度较高。
从植物激素信号转导(plant hormone signal transduction)、光合作用蛋白(photosynthesis-antenna proteins)、果糖和甘露糖代谢(fructose and mannose metabolism)等Pathway中候选出部分与吲哚-3-甘油磷酸合成酶、蛋白磷酸酶、己糖激酶、钙结合蛋白、叶绿素a/b结合蛋白有关的基因作为草地早熟禾干旱响应的候选基因。主要有c120656_g1、c122301_g3、c145664_g1、c117236_g1 、c119413_g2、c137695_g1、c123772_g1、c135567_g1等。
2.7 差异表达基因的qPT-PCR荧光定量验证
随机选取8个差异表达基因, 其中含有4个上调表达的差异基因(c111268_g1、c145664_g1、c145507_g1、c128115_g1)和4个下调表达的差异基因(c117236_g1、c119413_g2、c135104_g1、c93924_g1), 以Actin为内参进行qRT-PCR验证。由图5可知, 8个基因在干旱胁迫下的表达程度不同, 但表达趋势与高通量测序结果基本一致, 即表明测序结果真实可靠。

图5 差异表达基因的qRT-PCR验证Fig.5 Validation of DEGs data by qRT-PCR
3 讨论
植物在干旱胁迫下, 细胞感受干旱胁迫信号, 通过信号转导, 调控相关基因表达和生理反应, 同时从转录和翻译等不同水平做出响应。目前, 草地早熟禾尚未完成基因组测序, 没有完整的基因组序列可供参考, 因而不依赖基因组序列信息的高通量测序成为研究草地早熟禾干旱响应基因差异表达更好的选择。通过数据的统计评估可知数据质量良好。
在GO功能分析中, 生物学过程的差异表达基因差异在蛋白磷酸化、蛋白质修饰过程、磷代谢过程等;细胞组分显著差异在泛素连接酶复合物、后期促进复合物、细胞膜等;分子功能显著差异在转移酶、蛋白激酶、过氧化物酶、脂肪酸合酶等。这说明草地早熟禾干旱胁迫响应是一个复杂的生理变化过程, 要提高其抗旱性也不是单个基因可以完成的。许多研究表明, Ca2+参与细胞内信号转导过程。当植物受到胁迫时, 细胞质Ca2+浓度瞬时增加, 细胞质Ca2+可以与各种钙结合蛋白相互作用, 发挥相应的生物学功能。目前, 人们在植物中鉴定出钙调素、CDPKs(蛋白激酶)和CBLs(钙调磷酸酶类蛋白)这三类Ca2+感受体参与了胁迫信号转导[15-16]。有研究显示, 玉米丝氨酸/苏氨酸蛋白磷酸酶2C基因ZmPP2Ca与玉米对干旱胁迫的应答有关[17];紫花苜蓿促分裂原活化蛋白激酶MAPK基因与抗旱相关, 其中MMK4可能作为信号传递体参与磷酸化与去磷酸化的过程, 来调控紫花苜蓿对干旱的适应性反应[18];水稻OsDISI基因通过在转录水平上抑制一系列干旱正调控因子和诱导一系列干旱负调控因子的表达而负调控水稻的干旱胁迫响应过程[19]。本研究发现草地早熟禾干旱胁迫中蛋白激酶活性(GO: 0004672)上调基因394个与蛋白磷酸酶活性(GO: 0004721)上调基因14个都参与了草地早熟禾对干旱胁迫的响应。
在干旱胁迫下, 植物会产生内源激素来提高植物水分的利用效率。在这些激素中, 以ABA的研究最为广泛。ABA作为信号分子, 通过活化保卫细胞质膜上的PLC和Ca2+离子内流通道, 引起保卫细胞细胞质内Ca2+浓度的升高, 并激活K+外流通道, 最终使保卫细胞水势升高而失水, 引起气孔关闭, 抑制蒸腾作用, 从而增强植物对干旱的防御性反应。研究发现, 在植物体中许多基因的表达受干旱胁迫调控, 其中相当一部分基因响应ABA[20]。另外, 在对野生小麦根干旱响应蛋白的筛选与鉴定中发现, 表达差异蛋白主要涉及碳代谢等[21]。本研究中, 通过对草地早熟禾差异表达基因进行KEGG富集分析发现, 在前20显著性富集差异中, 与碳代谢(ko01200)、植物激素信号转导(ko04075)相关的基因相对较多。
在对拟南芥的耐受胁迫的研究中发现, 由茉莉酮酸介导的代谢途径的协调激活提供了对环境胁迫的抵抗力, 这些代谢途径中包含有吲哚-3-甘油磷酸合成酶的基因[22]。大麦穗干旱胁迫下发现, 它的芒中吲哚-3-甘油磷酸合成酶和叶绿素a/b结合蛋白基因下调表达[23]。在拟南芥中, 叶绿素a/b结合蛋白(LHCB)基因的下调表达会降低气孔运动对ABA的反应度, 从而导致拟南芥在干旱胁迫下的耐受性降低, 反之则增强[24]。在黑麦草的干旱胁迫研究中发现, 碳代谢相关基因有助于提高耐旱性, 其中己糖激酶(HxK)编码基因在抗性系中显示上调[25]。
使用qRT-PCR对随机选出的8个基因的表达量检测结果与高通量测序的表达结果总体趋势吻合, 但在具体表达倍数的数值上存在差异, 这可能与样本送测时间, 数据分析方法及检测灵敏度不同有关。
4 结论
本研究以草地早熟禾‘Nuglade’为材料, 以正常水分和干旱胁迫条件下的2个样本叶片进行转录组分析, 在转录水平上获得254331个基因的真实序列信息。检出17110个SSR位点, 通过差异基因筛选, 共鉴定出24465个差异表达基因。其中有12783个DGEs在干旱胁迫响应中表达上调, 11682个表达下调。分析得出, 与蛋白激酶、蛋白磷酸酶、碳代谢以及ABA等相关的基因可以作为研究草地早熟禾干旱响应机制的主要研究对象, 吲哚-3-甘油磷酸合成酶、蛋白磷酸酶、己糖激酶、钙结合蛋白、叶绿素a/b结合蛋白的基因或转录因子可作为与草地早熟禾干旱胁迫相关的应答候选基因, 从而揭示草地早熟禾叶片响应干旱胁迫的分子机制。
References:
[1] Li W. Cloning and Expression Analysis of Transcription Factors GenePpNACinPoapratensisL. Beijing: Chinese Academy of Forestry, 2011.
李伟. 草地早熟禾转录因子基因PpNAC的克隆和表达分析. 北京: 中国林业科学研究院, 2011.
[2] Xu L X, Han L B, Huang B R. Antioxidant enzyme qctivities and gene expression patterns in leaves of kentucky bluegrass in response to drought and post-drought recovery. Journal of The American Society for Horticultural Science, 2011, 136(4): 247-255.
[3] Xin J N. Genetic Transformation of Drought-resistance and Salt-resistance Gene in Kentucky Bluegrass (PoapratensisL.). Beijing: Beijing Forestry University, 2006.
信金娜. 草地早熟禾(PoapratensisL.)抗旱耐盐基因遗传转化. 北京: 北京林业大学, 2006.
[4] Zhang Z B, Shan L. Advances in inheritance of crop drought resistance physiological traits. Chinese Science Bulletin, 1998, (17): 1812-1817.
张正斌, 山仑. 作物抗旱生理性状遗传研究进展. 科学通报, 1998, (17): 1812-1817.
[5] Wang X C, Yang Z R, Wang M,etal. High-throughput sequencing technology and its application. China Biotechnology, 2012, (1): 109-114.
王兴春, 杨致荣, 王敏, 等. 高通量测序技术及其应用. 中国生物工程杂志, 2012, (1): 109-114.
[6] Duhoux A, Carrere S, Gouzy J,etal. RNA-Seq analysis of rye-grass transcriptomic response to an herbicide inhibiting acetolactate-synthase identifies transcripts linked to non-target-site-based resistance. Plant Molecular Biology, 2015, 87(4/5): 473-487.
[7] Zhou Q, Luo D, Ma L C,etal. Development and cross-species transferability of EST-SSR markers in siberian wildrye (ElymussibiricusL.) using illumina sequencing. Scientific Reports, 2016, 6: 20549.
[8] Zhu C, Ai L, Wang L,etal. De Novo Transcriptome Analysis of Rhizoctonia Solani AG1 IA Strain Early Invasion inZoysiajaponicaRoot. Frontiers in Microbiology, 2016, 7: 708.
[9] Ahn J H, Kim J, Kim S,etal. De novo transcriptome analysis to identify anthocyanin biosynthesis genes responsible for tissue-specific pigmentation inZoysiagrass (ZoysiajaponicaSteud.). Plos One, 2015, 10: e01379439.
[10] Zhao S, Zhang N, Liu D Y,etal. The impact of fenarimol application on the magnaporthe poae and fungi in the rhizosphere soil of Kentucky Bluegrass (PoapratensisL.). Journal of Nanjing Agricultural University, 2015, (4): 590-595.
赵爽, 张宁, 刘东阳, 等. 氯苯嘧啶醇施用对草坪斑枯病致病菌及根际土壤真菌的影响. 南京农业大学学报, 2015, (4): 590-595.
[11] Grabherr M G, Haas B J, Yassour M,etal. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 2011, 29(7): 130-644.
[12] Anders S, Wolfgang H. Differential expression analysis for sequence count data. Genome Biology, 2010, 11(10): 106.
[13] Young M D, Wakefield M J, Smyth G K,etal. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology, 2010, 11: 142.
[14] Kanehisa M, Araki M, Goto S,etal. KEGG for linking genomes to life and the environment. Nucleic Acids Research, 2008, 36(SI): 480-484.
[15] Zhang J J, Li J J, Nian H J. The role of calcium/calmodulin signaling pathways in the stresses: progress in researches. Chinese Journal of Microecology, 2013, (7): 858-860.
张晶晶, 李金金, 年洪娟. 钙/钙调素信号途径在胁迫中的作用研究进展. 中国微生态学杂志, 2013, (7): 858-860.
[16] Sanchez-Barrena M J, Fujii H, Angulo I,etal. The structure of the C-terminal domain of the protein kinase AtSOS2 bound to the calcium sensor AtSOS3. Molecular Cell, 2007, 26(3): 427-435.
[17] He L., Li F H, Sha L N,etal. Activity of serine/threonine protein phosphatase type-2C (PP2C) and its relationships to drought tolerance in maize. Acta Agronomica Sinica, 2008, (5): 899-903.
何亮, 李富华, 沙莉娜, 等. 玉米2C型丝氨酸/苏氨酸蛋白磷酸酶(PP2C)活性与耐旱性的关系. 作物学报, 2008, (5): 899-903.
[18] Bai X M. Cloning of MAPK gene which was activated by drought inMedicagosativa. Inner Mongolia Petrochemical Industry, 2011, (5): 18-20.
白雪梅. 紫花苜蓿(Medicagosativa)抗旱相关促分裂原活化蛋白激酶MAPK基因的克隆. 内蒙古石油化工, 2011, (5): 18-20.
[19] Ning Y S. Functional and Mechanistic Analysis of the SINA E3 Ligase OsDIS1 in Rice. Changsha: Hunan Agricultural University, 2011.
宁约瑟. 水稻SINA泛素连接酶OsDIS1的功能分析和作用机制研究. 长沙: 湖南农业大学, 2011.
[20] Kang Z L, Yang Y H, Zhang L J. Molecular mechanism of responsing to drought stress in plants. Journal of Maize Sciences, 2006, (2): 96-100.
康宗利, 杨玉红, 张立军. 植物响应干旱胁迫的分子机制. 玉米科学, 2006, (2): 96-100.
[21] Wang Y N, Liu C, Meng K,etal. Effects of exogenous carbon source on carbon and nitrogen metabolism of wheat under drought stress. Journal of Henan Agricultural Sciences, 2015, (10): 29-34.
王雅楠, 刘存, 孟珂, 等. 外源性碳源对干旱胁迫下小麦幼苗碳氮代谢的影响. 河南农业科学, 2015, (10): 29-34.
[22] Sasaki-Sekimoto Y, Taki N, Obayashi T,etal. Coordinated activation of metabolic pathways for antioxidants and defence compounds by jasmonates and their roles in stress tolerance inArabidopsis. Plant and Cell Physiology, 2006, 47S: 233.
[23] Abebe T, Melmaiee K, Berg V,etal. Drought response in the spikes of barley: gene expression in the lemma, palea, awn, and seed. Functional & Integrative Genomics, 2010, 10(2): 191-205.
[24] Xu Y, Liu R, Yan L,etal. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid inArabidopsis. Journal of Experimental Botany, 2012, 63(3); 1095-1106.
[25] Pan L, Zhang X Q, Wang J P,etal. Transcriptional profiles of drought-related genes in modulating metabolic processes and antioxidant defenses inLoliummultiflorum. Frontiers in Plant Science, 2016, 7: 519.
DifferentialgeneanalysisofPoapratensisinresponsetodroughtstress
LENG Nuan1, LIU Xiao-Wei2, ZHANG Na1, XU Li-Xin1*
1.InstituteofTurfgrassScience,BeijingForestryUniversity,Beijing100083,China; 2.NanjingOldMountainForestFarm,Nanjing211811,China
Drought is one of the main factors affecting the productivity ofPoapratensis. This study was undertaken to reveal the change of gene expression profile ofP.pratensisin the absence of a reference genome. The high-throughput Illumina Hiseq sequencing platform was used to investigate the transcriptome of a control group (CK) and a drought treated group (D) ofP.pratensis. The sequencing data were subsequently analyzed to help reveal the molecular mechanisms of drought response inP.pratensis. The results showed that 24465 differentially expressed genes were detected under drought treatment. After screening 4143 up-regulated genes and 4415 down-regulated genes were obtained, accounting for 34.98% of the total number of differentially expressed genes. The genes related to protein kinase, protein phosphatase, carbon metabolism and ABA can be used as the main research object to study the drought response mechanism ofP.pratensis. qRT-PCR analysis showed that the expression of 8 randomly differentially expressed genes was consistent with the high-throughput sequencing results. In addition, a series of genes for indole-3-glycerophosphate synthase, protein phosphatase, carbohydrate kinase, calcium binding protein and chlorophyll a/b binding protein were selected as candidate genes for drought stress related toP.pratensis. This laid the foundation for potentially revealing the molecular mechanism of drought response inP.pratensis.
Poapratensis; drought stress; high-throughput sequencing; transcription group
10.11686/cyxb2017130http//cyxb.lzu.edu.cn
冷暖, 刘晓巍, 张娜, 许立新. 草地早熟禾干旱胁迫转录组差异性分析. 草业学报, 2017, 26(12): 128-137.
LENG Nuan, LIU Xiao-Wei, ZHANG Na, XU Li-Xin. Differential gene analysis ofPoapratensisin response to drought stress. Acta Prataculturae Sinica, 2017, 26(12): 128-137.
2017-03-21;改回日期:2017-06-14
中国林学会——青年人才托举工程项目资助。
冷暖(1992-),女,山东威海人,在读硕士。E-mail:lengnuan99@163.com*通信作者Corresponding author. E-mail:lixinxu@bjfu.edu.cn
猜你喜欢
中国生殖健康(2020年4期)2021-01-18
幼儿100(2020年31期)2020-11-18
心电与循环(2020年1期)2020-02-27
疯狂英语·初中版(2019年4期)2019-09-10
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
小太阳画报(2018年6期)2018-05-14
江苏农业科学(2017年5期)2017-04-15
化学工业与工程(2015年1期)2015-02-10
湖北农业科学(2014年3期)2014-07-21
