
烟火药中Mg+H2O与MgO+H2O反应的竞争机理研究
2017-12-16 02:38陈永康陈明华邹永刚过乐驹
火工品 2017年5期
陈永康,陈明华,邹永刚,过乐驹
烟火药中Mg+H2O与MgO+H2O反应的竞争机理研究
陈永康1,陈明华2,邹永刚3,过乐驹1
(1.军械工程学院,河北 石家庄,050003;2.军械技术研究所,河北 石家庄,050003;3.齐齐哈尔北方华安集团公司,黑龙江 齐齐哈尔,161000)
为了明确氧化镁在含镁烟火药中作为安定剂的作用机理,对镁/水反应和氧化镁/水反应的竞争机理进行了研究。采用密度泛函B3LYP/6-311++G(d,p)方法优化反应路径中各驻点的几何构型,分析其电子结构并计算振动频率,确定过渡态和中间体,得到两种反应的反应势能曲线。对比发现氧化镁与水会先发生反应,在长贮中起到保护镁粉的作用。
烟火;镁/水反应;氧化镁/水反应;贮存;量子化学;密度泛函方法
镁作为燃料,由于其燃烧能够产生大量热量,发出耀眼白光,生成白烟等特点,已被大量应用于各种发光、发热、发烟的烟火剂中。在含镁烟火剂的生产过程中,综合考虑生产工艺和安全因素,烟火剂不可避免地会存在一定的水分。然而在贮存过程中,药剂中的水分会和镁发生反应,产生氢气,导致烟火药药柱的力学性能下降,包装鼓胀等。为解决这一问题,可将氧化镁作为安定剂加入到含镁的烟火药剂中。但氧化镁作为安定剂的作用机理尚不明确,对于镁和氧化镁与水发生反应的先后顺序仍有待确定。镁和水的反应较为简单,对该反应已有不少研究[1-5],最早Douglas等人[6]用基质隔离-分子光谱方法研究其反应机理;周星[7]对低温下镁和水的反应及其动力学进行了研究,通过试验研究了低温下镁水反应的反应进度,采用模式配合法确定反应机理函数并计算相关动力学参数。韩志江[8]采用从头算G2M(CC2)方法计算了镁和水在高温下的反应机理,其反应产物是氧化镁而非氢氧化镁,这和低温下的镁水反应是不同的[9]。而对于氧化镁和水的反应,目前有涉及氧化镁量子化学计算的相关研究[10-14],但针对氧化镁和水反应的理论计算相对较少,而且有关镁和氧化镁与水的竞争反应机理也未见报道。本文针对这一问题,利用量子化学方法,计算镁/水反应和氧化镁/水反应的机理,从理论上对氧化镁作为安定剂的作用机理进行解释。
1 反应机理
镁是活泼性很强的碱土金属元素,在低温下即可和水发生反应,生成氢氧化镁和氢气。其氧化物氧化镁也能与水发生反应,生成对应的氢氧化物即氢氧化镁。其反应机理如下:
Mg + 2H2O = Mg(OH)2+ H2
MgO + H2O = Mg(OH)2
本文对镁和水反应以及氧化镁和水反应的竞争机理进行了计算,优化两种反应中各反应物和产物的几何结构并计算振动频率,寻找过渡态和中间体,计算能量得到反应的势能曲线,最终确定两种反应的竞争关系。
2 理论计算
2.1 计算方法
所有的电子结构和能量计算均由Guassain 09程序包完成。采用密度泛函方法(Density Functional Theory,DFT)中的杂化泛函B3LYP方法,B3LYP方法属于Becke三参数杂化泛函,B3LYP使用 LYP 表达式提供的非局域关联,局域关联使用VWN 泛函III。在B3LYP/6-311++G(d,p)水平下优化各反应中反应物、产物和过渡态的几何结构并计算振动频率。对反应过渡态进行内禀反应坐标(IRC)计算对过渡态加以确认。对体系进行闭壳层计算。在B3LYP/ 6-311++G(d,p)水平下计算得到气态条件下两种反应的势能曲线。
2.2 结果与讨论
2.2.1 几何结构
在B3LYP/6-311++G(d,p)水平下优化各反应中各个驻点的几何构型,分析驻点的电子结构并计算振动频率。两种反应中各个驻点的优化几何结构如图1所示。得到相应的结构信息包括键长键角如表1所示。
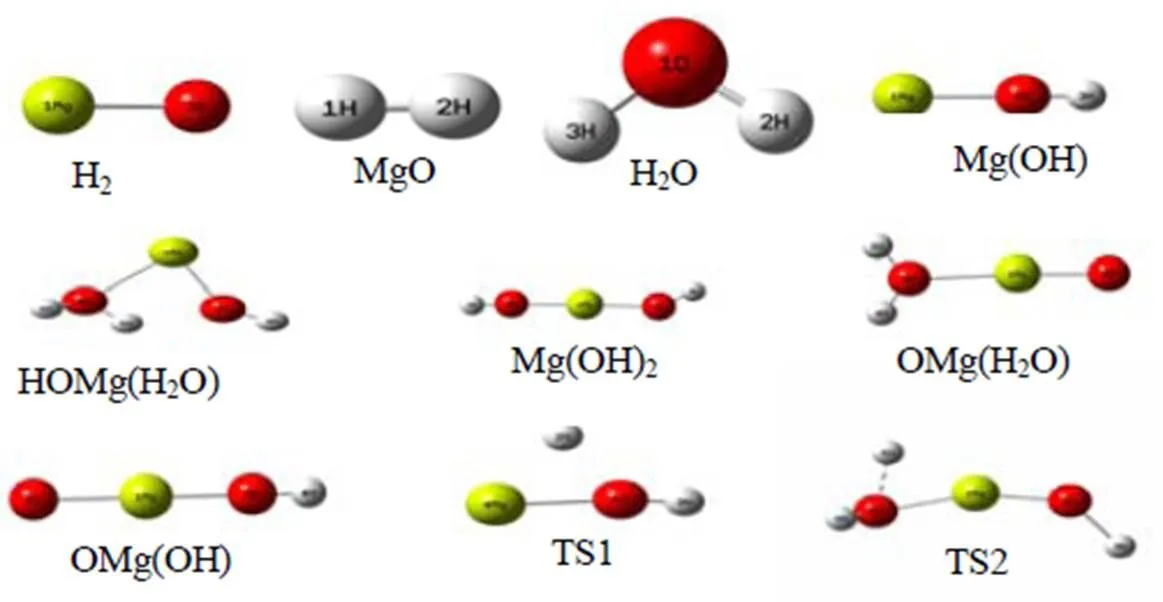
图1 各驻点的几何结构
表1 各驻点的结构参数

Tab.1 Geometric parameters of all stationary points
2.2.2 Mulliken重叠布居和偶极矩
计算得到各驻点的Mulliken重叠布居,结果如表2所示。根据键的Mulliken重叠布居数,可判断化学键的强弱。根据表2中的数据,总体而言Mg-O键的布居数在0.25~0.30之间,H-O键的布居数在0.3左右,相比而言Mg-O键要稍弱一些。同时,也有个别中间体在反应过程中形成的化学键不够稳定,布居数相对较小,如HOMg(H2O) 的Mg(1)-O(4)布居数为0.143 9,OMg(H2O)的Mg(1)-O(3)布居数为0.157 3。
表2 各驻点的Mulliken重叠布居

Tab.2 Mulliken populations of all stationary points
计算得到各驻点的偶极矩,结果如表3所示。
表3 各驻点的偶极矩
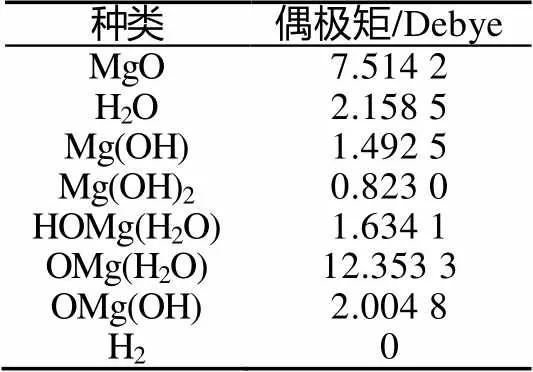
Tab.3 Dipole moments of all stationary points
分子的偶极矩是表征分子结构的重要参数,与分子的对称性和电荷分布状况有密切关系。反应产物Mg(OH)2的极性较小,偶极矩为0.823 0Debye,镁/水反应的两个中间体Mg(OH)、HOMg(H2O)偶极矩接近,分别为1.492 5Debye、1.634 1 Debye,氧化镁/水反应的中间体OMg(H2O)偶极矩较大,为12.353 3Debye。
2.2.3 振动频率
计算得到两种反应中各个驻点的频率,见表4。
表4 所有反应物、中间体、产物和过渡态的振动频率
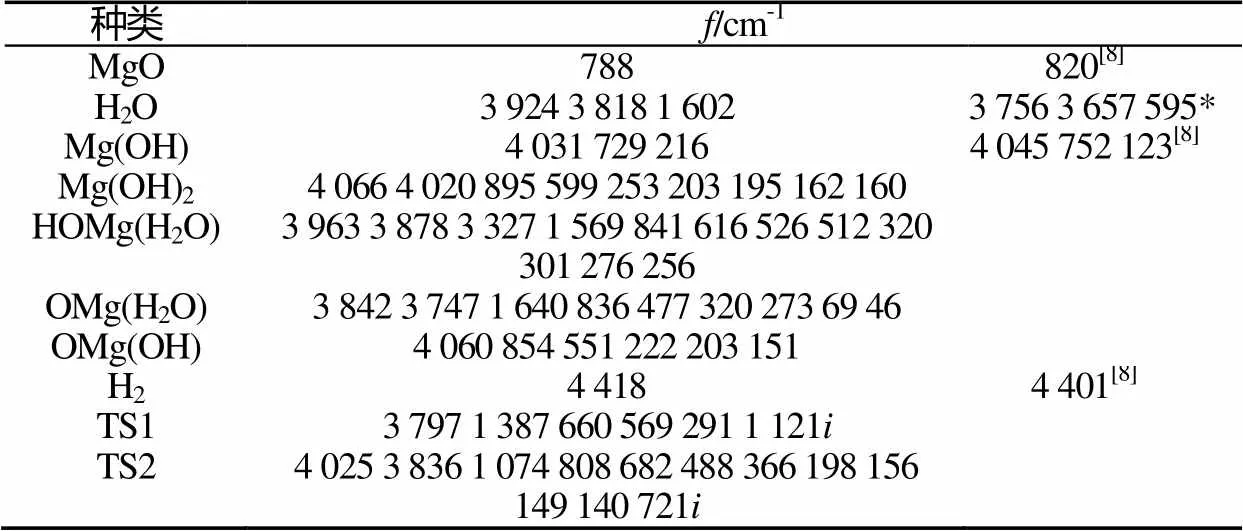
Tab.4 Vibrational frequencies of all reactants, transition states and products
注:*为NIST实验值。
由表4可以看出,过渡态TS1和TS2均有且仅有1个虚频。其中镁水反应中的TS1的虚频为1 121,TS2的虚频为 721。同时表4中列出相关的实验值和文献值,其中MgO伸缩振动频率为788cm-1,文献值为820cm-1。H2的伸缩振动频率为4 418 cm-1,文献值为4 401 cm-1。H2O的对称伸缩振动、反对称伸缩振动、弯曲振动频率分别为3 924 cm-1、3 818 cm-1、1 602 cm-1,实验值为3 756cm-1、3 657cm-1、595cm-1。本文的计算结果与实验值和文献值相吻合。
2.2.4 竞争机理
B3LYP/6-311++G(d,p)水平下计算得到两种反应中各驻点的结构能量、零点能(Zero-point energy)校正以及焓,结果见表5。
在B3LYP/6-311++G(d,p)水平下计算得到两种反应的反应势能曲线,如图2所示。结合势能曲线和结构信息,对反应机理进行分析。
表5 所有驻点的相对能量
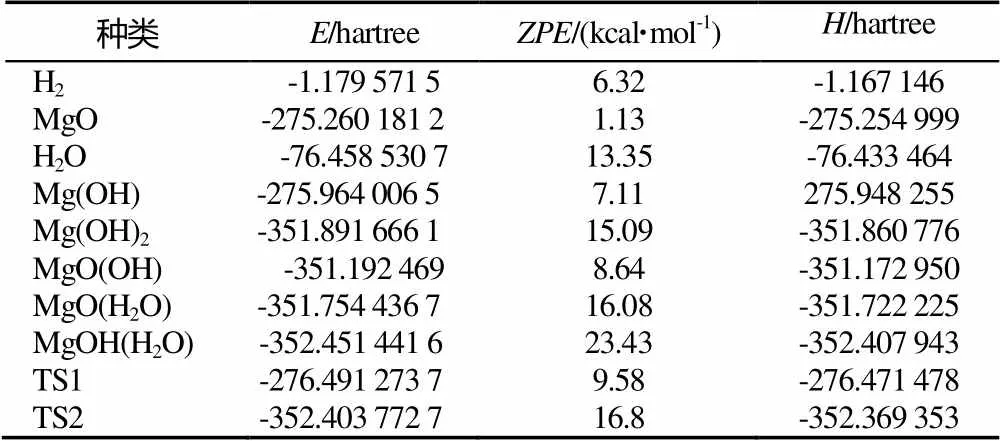
Tab.5 Energy values of all stationary points
镁/水反应:(1)Mg+H2O→Mg(OH)+H
Mg与水先经由过渡态TS1形成中间体Mg(OH),Mg原子接近水的O原子,O-H键逐渐拉长,水的O-H键长为0.961 2Å,过渡态TS1的O(1)-H(2)键键长为1.584 2Å,键明显拉长,而O(1)-H(3) 键的键长为0.966 2Å,也略有伸长。而后O(1)-H(2)断裂,脱去1个H原子。Mg原子和水O原子逐渐靠近,TS1的Mg(4)-O(1) 键键长为1.885 0Å,脱H形成中间体Mg(OH)后,其键长为1.799 2 Å,Mg-O键更加稳定。Mg(OH)的H(3)-Mg(1)-O(2)键角为179.88°,并非成一条直线。这一步反应的过渡态TS1能垒为37.98 kcal/ mol(158.76kJ/mol)。能垒相对较高,说明反应不易进行。
(2)Mg(OH)+H+H2O→Mg(OH)2+H2
中间体继续反应生成产物H2和Mg(OH)2,有两种反应通道:一种是直接形成最终产物,即Mg(OH)与1个水分子反应,水分子的O原子接近Mg原子,形成Mg(1)-O(3)键,其键长为1.7804 Å,另一个Mg(1)-O(2)键键长为1.798 2 Å,与Mg(OH)的相比都更短一些,说明更加稳定。参加反应的水分子的O-H键拉长断裂脱去1个H原子,最终形成H2和Mg(OH)2。产物Mg(OH)2几何结构与Mg(OH)类似,O(4)-Mg(1)-O(2) 和Mg(1)-O(2)-H(3)的键角都接近180°。这一步反应没有能量势垒。
Mg(OH)与H2O反应的另一个通道是先生成(HO)Mg(H2O),再生成最终产物Mg(OH)2+H2。
(3)Mg(OH)+H2O→(HO)Mg(H2O)
Mg(OH)先和水分子形成中间体(HO)Mg(H2O),其中(HO)Mg(H2O)的Mg(1)-O(2)和 Mg(1)-O(4)键长分别为1.894 2 Å、2.148 7 Å,Mg(1)-O(4)键相对较长,说明和水结合地不太牢固。由于水的加合,(HO)Mg (H2O)的O(3)-Mg(1)-O(2)键角为75.42°,这与Mg(OH)、Mg(OH)2的构型不太一样。同时根据表5 的计算结果,发现其相对能量较高,说明该物质并不稳定,容易进行下一步反应。
(4)(HO)Mg(H2O)+H→Mg(OH)2+H2
(HO)Mg(H2O)经由过渡态TS2生成最终产物Mg(OH)2+H2,TS2的两个Mg-O键的键长分别为1.771 3 Å、1.916 0 Å,比中间体(HO)Mg(H2O)的两个Mg-O键略短,与产物的Mg-O键长接近。说明Mg原子与两个O原子逐渐靠近,结构更加稳定。TS2的O(4)-H(6)键长为1.485 6 Å,键拉长断裂,脱去1个H原子形成最终产物Mg(OH)2+ H2,这一步反应的能垒为11.88 kcal/mol(49.7kJ/mol)。
氧化镁/水反应:(1)MgO+H2O→OMg(H2O)
氧化镁先与水分子形成中间体MgO(H2O),MgO(H2O)的Mg(1)-O(2)键长为1.764 9 Å,较MgO的1.762 7 Å略有伸长,中间体的Mg(1)-O(3)键长为2.099 2 Å,说明MgO与水结合地并不是很稳定,O(3)-Mg(1)-O(2)键角为176.80°。此过程没有能量势垒。
(2)OMg(H2O)→OMg(OH)+H
而后MgO(H2O)克服能垒形成中间体MgO(OH)和H原子,MgO(OH)的Mg(1)-O(2)键长为1.889 4 Å,Mg(1)-O(3) 键长为1.777 7 Å。与OMg(H2O)相比,Mg与两个O原子结合地更加牢固。MgO(H2O)中的H并不是以离子形式解离,因为通过能量计算,发现MgO(OH)-+ H+的能量为287.78 kcal/mol(1 202.92 kJ/mol),要远远高于MgO(OH)· + H·的15.05 kcal/mol(62.91kJ/mol),MgO(H2O)更倾向于生成MgO(OH)·+ H·。
(3)OMg(OH)+H→Mg(OH)2
最后中间体MgO(OH)与H原子形成最终产物Mg(OH)2。MgO(OH)的O(3)-Mg(1)-O(2)和O(3)-Mg (1)- H(4)键角均与Mg(OH)2的接近,其几何结构已经比较类似。
比较两种反应的势能曲线,氧化镁与水反应的能垒为15.05kcal/mol(62.91kJ/mol),而镁/水反应的能垒为37.98kcal/mol(158.76kJ/mol),得出结论是氧化镁先与水发生反应。
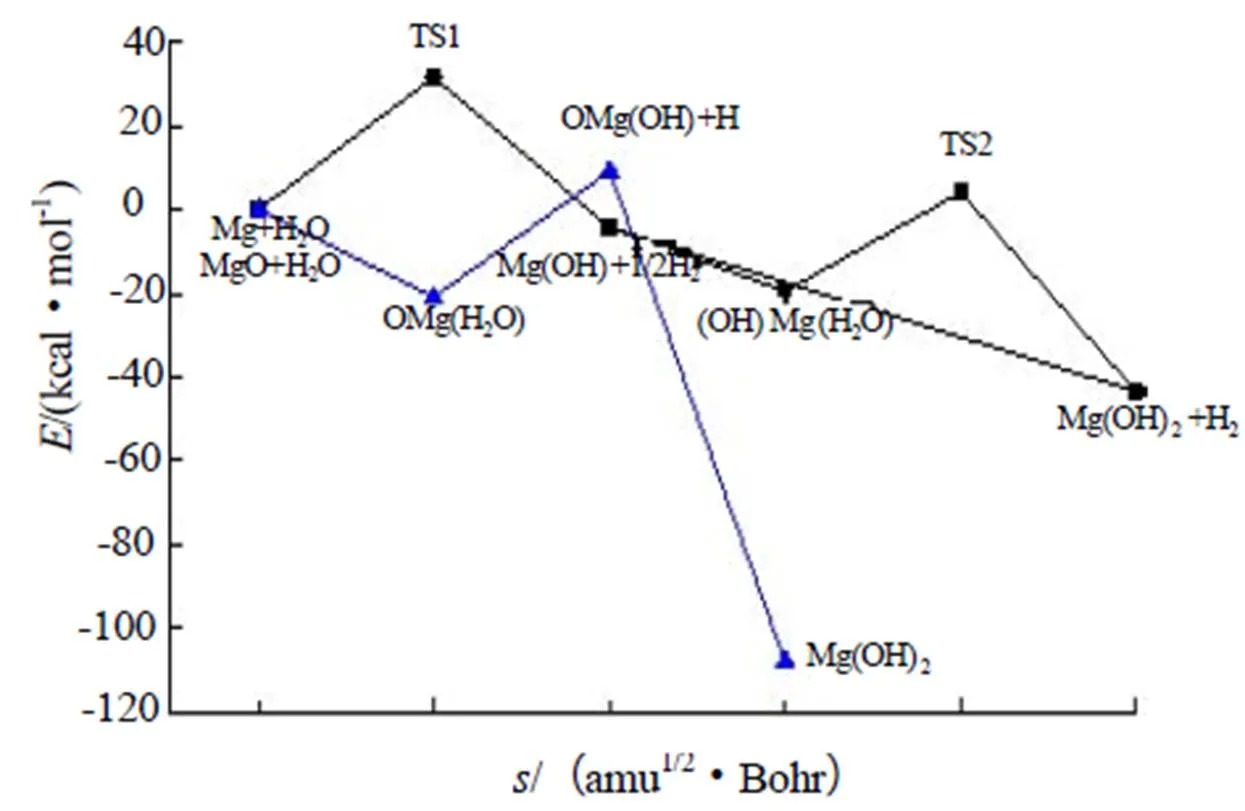
图2 两种反应的反应势能曲线
3 结论
本文对镁和氧化镁与水反应的竞争机理进行了理论计算,得出以下结论:(1)采用密度泛函方法优化反应路径中各驻点的几何构型,根据优化结果分析了驻点的电子结构,计算得到驻点的Mulliken 重叠布居数和偶极矩等。同时计算了振动频率,过渡态有且仅有1个虚频。计算结果与实验值、文献值相符。(2)得到各驻点的能量信息,对两种反应的反应机理进行分析,对比两种反应的反应势能曲线,镁/水反应的过渡态能垒为37.98kcal/mol(158.76kJ/mol),氧化镁/水反应的反应能垒为15.05kcal/mol(62.91kJ/mol),表明氧化镁与水的反应更容易进行。(3)结果表明氧化镁作为含镁烟火药剂中的安定剂,能够比镁先与药剂中的水分发生反应,生成氢氧化镁,起到中和水分,保护镁粉的作用。
[1] 陈静允. 钛-强碱性溶液、镁-水体系反应机制的研究[D]. 杭州:浙江工业大学, 2012.
[2] 厉雄峰. 镁/水体系反应行为及其副产物氢氧化镁吸附性能的研究[D]. 杭州:浙江工业大学, 2015.
[3] 杨栋, 张炜, 周星. 镁基水反应金属燃料与水反应模型及数值分析[J]. 推进技术, 2012, 33(1): 111-115.
[4] 周星. 镁基水反应金属燃料与水反应特性研究[D]. 长沙:国防科学技术大学, 2010.
[5] Sakai S. Theoretical studies of Mg(1S, 3P) atom reaction mechanisms with HF, H2O, NH3, HCl, H2S and PH3molecules[J]. Bull. Chem. Soc. Jpn., 1993(66): 3 326-3 333.
[6] Douglas M A, Hauge R H, Margrave J L. Electronic matrix isolation spectroscopic studies of the group IIA metal-water photochemistry[J]. High Temp. Sci., 1984(17): 201-206.
[7] 周星, 张炜, 郭洋,等.低温镁/水反应特性及反应动力学研究[J].固体火箭技术, 2011, 34(1): 71-75.
[8] 韩志江. 镁/水着火燃烧模型及高温均相反应机理研究[D]. 杭州:浙江大学, 2012.
[9] 周星,张炜,李是良.镁粉的高温水反应特性研究[J].固体火箭技术, 2009, 32(3):302-305.
[10] Yoshimine M. Computed ground state properties of BeO, MgO, CaO and SrO in moleculer orbital approximation[J]. Journal of the Physical Society of Japan, 1968, 78(4): 1 100-1 119.
[11] Bunker P R, Kolbuszewski M, Jensen P, et al. New rovibrational data for MgOH and MgOD and the internuclear potential function of the ground electronic state[J]. Chemical Physics Letter, 1995, 239(4-6): 217-222.
[12] Zou M S, Guo X Y, Huang H T. Preparation and characterization of hydro-reactive Mg-AI mechanical alloy materials for hydrogen production in sea water[J]. Journal of Power Source,2012(19): 60-64.
[13] Jain S K, Rout C, Rastogi R C. Density functional study of the isomerisation of MOH(M=Be and Mg)[J]. Chemical Physics Letter,2000, 311(5-6): 547-552.
[14] 赵臻,王娣,汪琦. 碳酸镁热分解主要反应通道的量子化学分析[J]. 辽宁科技大学学报, 2014, 37(2): 119-125.
Study on Competition Mechanism of Mg+H2O and MgO+H2O Reaction in Pyrotechnics
CHEN Yong-kang1,CHEN Ming-hua2,ZOU Yong-gang3,GUO Le-ju1
(1.Ordnance Engineering College,Shijiazhuang,050003;2.Ordnance Technology Research Institute,Shijiazhuang,050003;3.Northern Hua’an Group Company,Qiqiha’er,161000)
In order to determine the mechanism of magnesia used as stabilizing agent in pyrotechnic composition containing magnesium powder, magnesium/water reaction and magnesia/water reaction mechanism were investigated theoretically. The optimized geometries and frequencies of the stationary points of two reactions were calculated at B3LYP/6-311++G(d,p) level. The transition state and intermediate were confirmed, the potential energy curves of two reactions were also obtained. It is shown that magnesia will react with water firstly, so the magnesium would be protected during storage.
Pyrotechnics;Magnesium/water reaction;Magnesia/water reaction;Storage;Quantum chemistry;Density functional method
1003-1480(2017)05-0031-05
TQ567
A
10.3969/j.issn.1003-1480.2017.05.009
2017-07-01
陈永康(1991 -),男,在读博士研究生,主要从事弹药保障与安全研究。
江苏省研究生培养创新工程项目(SJLX15_0145)
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
原子与分子物理学报(2022年3期)2022-03-05
无机盐工业(2022年1期)2022-01-19
辽宁科技大学学报(2021年2期)2021-07-22
科技视界(2021年18期)2021-07-14
汕头大学学报(自然科学版)(2020年4期)2020-12-14
世界有色金属(2020年17期)2020-11-28
无机盐工业(2019年7期)2019-12-27
青岛大学学报(工程技术版)(2019年2期)2019-09-10
山东工业技术(2018年9期)2018-05-26
