
抗心律失常药丙吡胺的分光光度测定新法
2017-12-14 01:35
分析仪器 2017年6期
(四川大学华西药学院,成都 610041)
抗心律失常药丙吡胺的分光光度测定新法
方梅龙雯
(四川大学华西药学院,成都 610041)
建立了一种用于抗心律失常药丙吡胺的分光光度测定新法。此法基于下述4个步骤:(1)丙吡胺分子中的氮原子能与过量四苯硼钠(Ph4B-Na+)定量反应生成沉淀;(2)滤液中余量的四苯硼钠(Ph4B-Na+)负一价大阴离子(Ph4B-)与Cu+-2,9-二甲基-1,10-菲罗啉(正一价大阳离子)螯合物生成缔合物;(3)这黄色的缔合物经异戊醇定量地萃取;(4)在最大吸收波长470纳米处相对于参比溶液测定吸光度。在最佳条件下,校正曲线的线性关系良好,线性回归系数R2=0.9991,遵守比尔定律的浓度范围是0~2×10-4mol/L的, 丙吡胺。此法用于丙吡胺模拟注射液的测定,其标准加入的回收率范围为99.76%~100.66%,日内和日间变异系数分别是0.25% 和0.42%。测定3×10-4mmol的 Ph4B-时,≦ 2×10-4mmol量的Na+,K+,Ag+,Mg2+,Zn2+,Co2+,Cd2+,Al3+及≦ 5×10-5mmol量的Ca2+,Sr2+,Ba2+,Fe2+,Fe3+和≦ 5×10-4mmol量的Cl-,NO3-不干扰。而5×10-5mmol量的Cu2+能抢夺Cu+-2,9-二甲基-1,10-菲罗啉螯合物中的2,9-二甲基-1,10-菲罗啉,从而有干扰,但加入盐酸羟胺将Cu2+还原成Cu+,即可消除此干扰。本法简便,可靠,未见国内外报道。
抗心律失常药 丙吡胺(对映体混合物) 分光光度测定法、四苯硼钠 2,9-二甲基-1,10-菲罗啉 缔合物 溶剂萃取
丙吡胺(Disopyramide),别名双异丙吡胺、吡二丙胺、异脉停、达舒平、诺佩斯(Norpace)、Pythmodan,属IA类抗心律失常药,钠离子通道阻断剂(抑制剂),即使钠离子通道狭窄或关闭,阻止钠内流。丙吡胺是广谱抗心律失常药,比奎尼丁(Qninidine)用途广,且副作用小。可替代最早用于临床的奎尼丁和普鲁卡因胺(Procainamide),用于各型早搏,各型心动过速、房颤、房搏等,对伴有某些症候群的心动过速疗效尤佳[1]。
丙吡胺的测定,可用高效液相色谱法[2-11]、传感器[12,13]毛细管电动色谱[14]等,但高效液相色谱仪价格相对贵许多,根据需要,试用和更换不同色谱柱也并非十分简便,作为流动相的有机溶剂用量较大。毛细管电泳法更方便,用途也广[15-17],但毛细管电泳仪价也贵,且用于分离丙吡胺的对映体更能体现其长处[18-23]。对于丙吡胺总量(对映体混合物)的测定,我们认为分光光度法是最方便的方法之一。因此,新建分光光度法测定丙吡胺总量的方法很必要。但丙吡胺分子中无发色团,不能在可见光区测,只能在紫外光区测,但紫外分光光度计价格相对于可见区分光光度计昂贵,且在紫外区测定所受的干扰成分(包括各种溶剂、杂质等)很多。因此,制定一种能在可见光区测定它的方法是既有实用意义(各种测定方法各有长处和短处及其最佳适用范围。给相关工作者提供可视实际需要选用的多种方法中之又一种,不断丰富科技文献宝库,是广受欢迎的),又有学术意义(如何把看似不可能的状况转变为可能?而欲实现这种转变,需设计和探索出哪些切实的歩骤? 建成的方法用于样品分析,考察其效果是否令人较为满意?)的工作。本文即旨在于此。
如何建立一种在可见光区,间接分光光度测定无色药物丙吡胺的新体系呢?这要作者随时广泛掌握分析科学发展动态,知己知彼,并调用多种知识且将它们综合运用,以解决指定难题。
高鸿院士曾带领他的研究团队,用四苯硼钠标准溶液直接示波滴定多种分子中含氮的药物,并将其专著寄赠我们两本[24-26]。
我们从中颇受启发,并看出三点: (1) 四苯硼钠可能与所有含氮药物反应生成沉淀;(2)四苯硼钠标准溶液与含氮药物的反应是定量进行的,否则,不能在滴定终点时按四苯硼钠的消耗量以当量定律计算含氮药物的含量;(3) 四苯硼钠标准溶液与含氮药物的反应是瞬间进行的,否则,不能直接滴定而只能回滴或代滴。
据此, 我们推测四苯硼钠标准溶液还可能与尚未报道过的其它含氮药物(例如我们正在研究的丙吡胺)发生定量和快速的反应。
若简单地将四苯硼钠标准溶液直接示波滴定含氮药物含氮药物,应是很易成功之事,仅增加将四苯硼钠标准溶液直接示波滴定又一含氮药物而已。
但我们的目的是只想借鉴四苯硼钠可能与丙吡胺进行瞬间,定量的反应,却不用滴定方式完成测定,而设计出以间接分光光度的方式完成含氮药物丙吡胺的测定。增添一类可供有关人员酌情选用的新方法。为此,可向丙吡胺待测试液中加入定量过量的四苯硼钠溶液, 瞬间定量反应完成后,分光光度测定剩余四苯硼钠。由于四苯硼钠与丙吡胺一样,分子中不含发色团,也是无色化合物,而紫外分光光度测定受干扰的情况多,因此,需建立可见区分光光度测定法。这关键是让无色的四苯硼钠 (在其水溶液中电离为四苯硼阴离子和钠离子)转变成有色化合物,而希望这种有色化合物也能瞬间,定量生成,且其生色分子的截面积尽量大,使测定的灵敏度尽量高。要达此目的,就让剩余的四苯硼阴离子与有色的螯合物(比一般配合物稳定得多)大阳离子瞬间,定量地缔合成有色的大缔合物。再将此大缔合物溶解于适宜的有机溶剂或分散于胶束介质中,成为均匀透明的体系,以利于分光光度准确测定。
1 仪器与试剂
1.1 仪器
721分光光度计,四川分析仪器厂出品。
1.2 试剂
(1)丙吡胺标准溶液: 准确称取丙吡胺固体(美国Sigma公司出品)7.30mg,用去离子水溶解并定容至50mL,浓度为3.34×10-4mol/L。
(2)四苯硼钠标准溶液: 准确称取四苯硼钠固体56.0mg,用20mL去离子水溶解。称取硝酸铝固体0.3g,溶于20mL去离子水,用0.1 mol/L氢氧化钠溶液调节pH至8~9,将此胶状液体倒入此四苯硼钠溶液中,过滤,将此滤液调节pH至8~9,用去离子水定容至100mL,用标准硝酸银溶液标定,得到四苯硼钠标准溶液,其浓度为3.13×10-3mol/L。
(3)硫酸铜标准溶液: 准确称取CuSO4.5H2O固体102.10mg,用加有2mL0.1 mol/L稀盐酸溶液的去离子水溶解并用水定容至100mL,Cu2+浓度为4.09×10-3mol/L。
(4)硫酸高铁銨标准溶液: 准确称取硫酸高铁銨固体1.0038g,置于烧杯中,加入浓硫酸5.0mL溶解,移入已有30mL去离子水的100mL容量瓶中,摇匀后,从容量瓶中倒出少许溶液洗涤烧杯中残留物,重复一次,倒回容量瓶中,再用去离子水稀释至容量瓶刻度,摇匀。
(5)1,10-菲罗啉溶液;0.15%溶液。
(6)2,9-二甲基-1,10-菲罗啉溶液: 称取2,9-二甲基-1,10-菲罗啉固体67.30mg,用5%稀盐酸溶液溶解后,用去离子水定容至50mL,浓度为6.20 ×10-3mol/L。
(7)2,2-联吡啶无水乙醇溶液: 称取2,2-联吡啶固体678.5毫克,用无水乙醇溶解并定容至50毫升,浓度为 8.688×10-2mol/L。
(8)盐酸羟胺溶液: ①5%溶液;②称取盐酸羟胺固体693.0mg,用去离子水溶解并定容至50mL,浓度为 1.995×10-1mol/L。
(9)抗坏血酸溶液: 5%。
(10)二苯硫巴腙(双硫腙)储备溶液: 称取双硫腙固体约50mg,用无水乙醇溶解并定容至50mL,浓度为约0.1%。
(11)醋酸-醋酸钠缓冲溶液: 将0.2 mol/L的醋酸与0.2 mol/L的醋酸钠溶液,按1:4的体积比混合,制得pH约5.8的缓冲溶液,用去离子水溶解100mL,摇匀。
(12)氯化钠注射溶液。
(13)阿拉伯树胶溶液: 1%。
(14)吐温-80溶液: 5%。
(15)十六烷基三甲基溴化胺溶液: 1%。
(16)曲拉通溶液: 10%。
(17)乙酸乙酯;正丁醇;异丁醇;异戊醇;氯仿。
2 结果与讨论
2.1 实验过程
准确吸取丙吡胺标准溶液进入,加入过量、定量的四苯硼钠标准溶液,与其反应生成沉淀,干过滤或湿过滤或离心沉降,分离并洗涤此沉淀,洗涤液并入滤液中,向滤液中加入铜(II)或铁(III)、还原剂盐酸羟胺或抗坏血酸、N,N-螯合剂(生成溶于水有色的螯合物大阳离子),使其与剩余的四苯硼钠阴离子生成不溶于水,而溶于有机溶剂的有色缔合物沉淀。加入季铵盐表面活性剂使缔合物沉淀分散成均匀的胶束溶液或加入有机溶剂萃取,使缔合物沉淀溶解入有机相中,定容。倒入吸收池中,相对于参比溶液,在有色缔合物溶液的最大吸收波长处,测定吸光度。
2.2 实验条件的优化
由于本法基于待测成分丙吡胺与过量定量的四苯硼阴离子反应后,测定剩余四苯硼阴离子含量,而为要测剩余四苯硼阴离子含量,是利用四苯硼阴离子与螯合物大阳离子瞬间定量生成有色缔合物后,分光光度测定所生成此缔合物的含量。而测此缔合物时的灵敏度和选择性越高越好。因此,就需优选几方面的条件。包括:
(1)螯合物大阳离子的选择;
(2)有机溶剂或表面活性剂的选择:为便于分光光度法(只适于)测定透明而均匀的体系,就要考虑如何将缔合物溶解或分散为均匀体系,即选择有机溶剂(溶解)萃取方式还是选择胶束增溶(分散)?就存在选择什么有机溶剂,或选择什么表面活性剂.以灵敏度,选择性,再现性,准确性,简便性几方面为标准作选择标准的问题。
以下便是这一系列试验的情况.为提高选择的效率,先作简便的、初步的定性选择,在此基础上大幅度减小了进行定量选择的工作量。
2.2.1 以定性试验来选择
本法中所测定的缔合物,是由与待测成分丙吡胺反应后剩余的四苯硼阴离子与有色的螯合物大阳离子生成仍然有色物质。
四苯硼盐的选择:常见四苯硼盐有钠、钾、铷、铯、铊、铵、银等盐,其中,四苯硼钠在水中溶解度较大(8.8×10-1mol/L)。而它的其他一价金属盐水中溶解度皆小,小3~4个数量级(×10-4mol/L~10-3mol/L),因此,四苯硼钠溶液是测定其他一价金属的好试剂。也是沉淀含氮有机化合物季铵盐(生成白色沉淀)的好试剂[3]。但尚无人用它测定丙吡胺。四苯硼钠在水中完全电离为四苯硼阴离子和钠离子。设计本方案时就先指定用它,所以,就不用通过实验来选择了,丙吡胺是一分子含三个氮原子的有机化合物[1],本项目就研究四苯硼钠在测定丙吡胺中的应用。
上述有色物质并不是孤立的,而是存在于显色体系中。所以,显色体系包括:一定价数的金属阳离子(若用高价金属阳离子,则还要同时用还原剂)、螯合剂、四苯硼阴离子)的选择是决定性的。
而螯合物大阳离子由中性螯合剂与金属阳离子(也称为中心离子)两部分构成。所以,螯合物大阳离子的选择,就包括:既要选中性螯合剂(以便它与金属阳离子生成带正电荷的螯合物大阳离子,后者才与剩余四苯硼阴离子瞬间定量生成有色的缔合物,从而分光光度测定此有色的缔合物量,据以计算剩余四苯硼阴离子量。由定量过量加入的四苯硼阴离子总量减去剩余四苯硼阴离子量,即得到与丙吡胺定量反应的四苯硼阴离子量。从而按丙吡胺与四苯硼阴离子反应的比例算出样品中的丙吡胺含量。实验后来设计出直接查丙吡胺含量的工作曲线,而不需繁琐的计算。)又要选金属阳离子。
(1)中性螯合剂的选择
世界著名的乌克兰分析化学家巴布科曾提出: 无色金属离子与无色络合剂反应,生成无色络合物;无色金属离子与有色络合剂反应,生成有色络合物;有色金属离子与无色络合剂反应,生成有色络合物;有色金属离子与有色络合剂反应,生成有色络合物。
1,10-菲罗啉类是很好的中性螯合剂,能与有色的金属阳离子生成有色的、稳定的螯合物大阳离子,后者很易与四苯硼阴离子瞬间定量生成有色的缔合物沉淀。但现还市售有它的类似物2,9-二甲基-1,10-菲罗啉。及其它类似物2,2’-联吡啶、双硫腙。在本研究中对它们的效果进行比较是有益的。
1,10-菲罗啉与2,9-二甲基-1,10-菲罗啉两种中性螯合剂的比较,2,9-二甲基向母体上的加入,提高了选择性,也增大了分子的摩尔质量,使四苯硼阴离子缔合更完全。结果表明:用2,9-二甲基-1,10-菲罗啉为此处的中性螯合剂比所试其它螯合剂更好。
(2)中心离子的选择
Fe3+、Fe2+、 Cu2+、Cu+均是可被1,10-菲罗啉类和2,2’-联吡啶类中性螯合剂稳定螯合的中心离子。在本研究中对它们的效果进行比较也是有益的。实验分别以它们为中心离子,与1,10-菲罗啉、2,9-二甲基-1,10-菲罗啉、2,2’-联吡啶、双硫腙组成9种显色体系。结果表明:Cu+为中心离子更好。
(3)还原剂的选择
抗坏血酸与盐酸羟胺的比较:结果表明:以乙酸乙酯萃取时,用抗坏血酸和盐酸羟胺作还原剂,均可使螯合物大阳离子上层和下层为黄色透明,不带电的有色缔合物上层为黄色透明,下层为无色透明,效果最不好。用正丁醇、或异丁醇萃取时,用抗坏血酸和盐酸羟胺作还原剂,均可使螯合物大阳离子上层为无色透明,下层为黄色透明,不带电的有色缔合物上层为痕黄色透明,下层为无色透明。异戊醇萃取时,用抗坏血酸和盐酸羟胺作还原剂,均可使螯合物大阳离子上层为无色透明,下层为黄色透明,而不带电的有色缔合物上层为黄色透明,下层为无色透明。所以,用异戊醇萃取最好。但用抗坏血酸作还原剂不如用盐酸羟胺作还原剂好,因有机层为半透明,使测得到吸光度比实际值高,且抗坏血酸在空气中很不稳定,极易被氧化变黄。故选用盐酸羟胺作还原剂。
(4)缓冲溶液使用的必要性
为避免金属离子水解为氢氧化物类沉淀,也避免中性螯合剂有效浓度发生变化,此螯合反应宜在近中性的醋酸-醋酸钠缓冲溶液中进行,不宜在任何介质中进行。
总起来比较,以Cu2+-盐酸羟胺-2,9-二甲基-1,10-菲罗啉-醋酸-醋酸钠为选定的显色体系,
(5)反应物的加入量
为保证待测的丙吡胺被完全反应,在Cu2+-盐酸羟胺-2,9-二甲基-1,10-菲罗啉-醋酸-醋酸钠-四苯硼钠显色体系中,盐酸羟胺-2,9-二甲基-1,10-菲罗啉是过量的,保证Cu2+被完全反应,有良好的线性关系。它们对于剩余的四苯硼钠又都是足够过量的,保证剩余的四苯硼钠完全反应。而剩余的四苯硼钠又对于待测的丙吡胺是足够过量的,以保证待测的丙吡胺被完全反应生成缔合物。所测得的结果才能反映丙吡胺的含量。经试验和计算,确定测定时应加入过量的各反应物Cu2+、盐酸羟胺、2,9-二甲基-1,10-菲罗啉、四苯硼钠的摩尔数比为1∶8∶10∶0.5。
2.2.2 以定量试验来优选测定条件
2.2.2.1 胶束增溶与溶剂萃取的比较
基于螯合物大阳离子是带电荷,可溶于水,而不溶于极性弱的有机溶剂中的;不带电的有色缔合物则是不溶于水,而溶于极性弱的有机溶剂中的;借此,用某种有机溶剂萃取,可望将不带电的有色缔合物与余量的带电荷螯合物大阳离子分离(希望不带电的有色缔合物完全进入有机溶剂,而希望余量的带电荷螯合物大阳离子完全留在水层中),避免后者对测定前者时的干扰。
另一方面,表面活性剂却可将不溶于水的沉淀分散为均匀透明的胶状体系。操作比有机溶剂萃取更简单,但表面活性剂在将不溶于水的有色缔合物沉淀分散为均匀透明的胶状体系(显色试液)时,也使余量的带电荷螯合大阳离子(应仅存在于空白溶液中,但却有部分留在显色试液中)存在于同一胶状体系中,即不易将余量的带电荷螯合物大阳离子与不带电的有色缔合物截然分开,造成显色试液与空白溶液颜色区别小,而对有色缔合物的光度测定造成干扰。
为检验这两方式哪种实际效果,实验试用了阳离子表面活性剂十六烷基三甲基溴化胺、非离子表面活性剂阿拉伯树胶溶液、吐温-80溶液曲拉通溶液几种表面活性剂来分散有色缔合物和余量的带电荷螯合物大阳离子,以及乙酸乙酯、正丁醇、异丁醇、异戊醇、氯仿几种极性不同的有机溶剂来萃取有色缔合物。结果表明:用所试任何表面活性剂效果都不如用所试有机溶剂萃取好。进一步考察了所试几种有机溶剂的效果,介电常数:异戊醇14.7;乙酸乙酯6.02、正丁醇17.1、异丁醇17.7、氯仿4.8,结果表明异戊醇沸点较高,难挥发,在吸收池中测定时,不必加盖萃取有色缔合物效果最好,与水分层最清楚,萃取层颜色也最稳定,且与氯仿相比,无毒。基于此本实验未对氯仿效果进行实验。
2.2.2.2 四苯硼钠溶液配制方法的比较
本研究中四苯硼钠起非常重要的作用,但它在水中容易水解,故其水溶液很不稳定。经查阅聊文献,配制方法有以下4种:
(1)称取四苯硼钠固体,直接溶于去离子水中。
(2)称取四苯硼钠固体,加入少量氢氧化钠和去离子水,移入容量瓶中定容。
(3)称取四苯硼钠固体,用少量去离子水溶解,加入定量新鲜的氢氧化铝,搅拌1分钟,放置片刻,过滤,滤液用0.1N氢氧化钠调节pH至8~9,加水稀释定容。
(4)称取四苯硼钠固体7.0g,加50mL去离子水,振摇使溶解。加入新配制的氢氧化铝凝胶,(取三氯化铝1.0g,溶于25mL水中,在不断搅拌下,缓缓滴加氢氧化钠溶液调节pH至8~9),加氯化钠16.6g,充分搅匀,加水250mL,振摇15分钟,静置10分钟,过滤,滤液中滴加氢氧化钠溶液至pH为8~9,再加水稀释至1000mL,摇匀。此法为药典所载方法。
在连续几个时间点,用硝酸银标准溶液标定对用以上4种配制方法所制得的四苯硼钠溶液的稳定性进行考察,结果表明上述(3),(4)两种方法所得四苯硼钠溶液的稳定性较好。从配制手续简便上考虑,实验选用方法(4)配制,然后用硝酸银标准溶液标定其浓度。
2.2.2.3 缔合物沉淀过滤方法的比较
文献[2、3、4]中对于四苯硼钠与待测含氮化合物反应生成的沉淀,用干过滤方法(个别地方用吸滤,见该书282页)处理,即等浓度过滤,再分别量取一定体积的滤液迳行后续实验,而我们采取湿过滤,即等量过滤过滤,保证了剩余的四苯硼钠全都进入滤液中,参与后续实验,两法各有优缺点。干过滤方法优点是能更好地保证各管的平行操作,且参加反应的滤液体积较小,用于本项测定时,其缺点是因为每管中剩余四苯硼钠的浓度不同,在量取滤液时,就得使用对应的移液管,不能混淆,故操作较麻烦,为此采取的湿过滤法直接将全部滤液用于实验,操作较简便、快速。但为保证剩余四苯硼钠完全进入滤液,虑完溶液后,就要用定量2.00mL去离子水,冲洗滤纸及试管,这样就增大了反应液的体积,增加了后面萃取的麻烦。
2.2.2.4 溶剂萃取时间与萃取程度的关系
对以前的文献报道中,不管其萃取是否完成,实验仅就所选定的体系在其最佳条件下考察萃取时间与萃取程度的关系。平行作5份,每份的萃取(振摇)时间不同。振摇频率一律保持每秒2次。结果见表1。

表1 萃取时间与萃取程度的关系
可见,萃取20 至30秒吸光度值已差别不大,即振摇20秒已萃取完全。
2.2.2.5 吸光度与测定时间的关系
加入4.00mL异丁醇萃取有色缔合物沉淀,以每秒振摇2次的频率振摇20秒,使缔合物沉淀完全溶入有机相中。立即将此异丁醇有机相吸入吸收池中,相对于参比溶液,在有色缔合物异丁醇溶液的最大吸收波长470nm处,于不同时间进行吸光度的测定,结果见表2。

表2 吸光度(470纳米处)随时间的变化
可见,异丁醇萃取后,可在3分钟至1小时内测定吸光度,相对误差小于3%。
2.2.2.6 吸收曲线的绘制
对溶剂萃取与胶束增溶分别绘制其最佳条件下波长范围450~700nm的吸收曲线,对它们的最大吸收波长、对比度、摩尔吸收系数进行比较。最后选定的体系最大吸收波长为470nm,摩尔吸收系数3.86×10-3L·mol-1(图1)。

(其余各条胶束增溶的吸收曲线、溶剂萃取的吸收曲线在此处从略)图1 Cu(I)-2,9-二甲基-1,10菲罗啉-异戊醇吸收曲线
2.2.2.7 工作曲线的绘制
(1)测剩余四苯硼阴离子用的工作曲线的绘制:分别绘制了检测四苯硼阴离子的4种显色体系的工作曲线。据以比较它们的线性关系范围.以便从中选择1种最佳显色体系,最后选中2,9-二甲基-1,10-菲罗啉-Cu2+-盐酸羟胺-四苯硼阴离子显色体系,在此最佳显色体系下才绘制用于测丙吡胺的工作曲线。
(2)测丙吡胺用的工作曲线的绘制:用2,9-二甲基-1,10-菲罗啉-Cu2+-盐酸羟胺-四苯硼阴离子显色体系显色后,用异戊醇萃取,将此不含丙吡胺的萃取液作测定对象(颜色最深),而依次用含不同量丙吡胺的异戊醇萃取液分别作参比(颜色依次变浅),在最大吸收波长测吸光度。
线性回归系数R2=0.9991
这样,可由此丙吡胺工作曲线,直接查得试液中丙吡胺含量。而不必从加入的四苯硼阴离子总量减去剩余四苯硼阴离子量(由这样检测的吸光度与剩余四苯硼阴离子量成正比。由此工作曲线查出剩余四苯硼阴离子量)的差,来计算丙吡胺含量
2.2.2.8 样品测定
准确吸取丙吡胺模拟溶液,加入过量、定量的四苯硼钠标准溶液,与丙吡胺反应生成沉淀,湿过滤,分离并用去离子水充分洗涤此沉淀,将全部洗涤液放在20mL比色管中并入滤液。向此滤液中加入0. 5020mL4.09×10-3mol/L的Cu2+标准溶液、0. 5020mL5%盐酸羟胺溶液,0. 5020mL醋酸-醋酸钠缓冲溶液,摇匀,放置1min,使Cu2+完全还原成Cu+,加入1.0020mL6.20 ×10-3mol/L的2,9-二甲基-1,10-菲罗啉溶液(生成橙红色螯合物大阳离子),使其与过量(剩余)的四苯硼阴离子生成有色的缔合物沉淀。仔细加入去离子水调节液面恰至1020mL刻度线处定容。加入4.0020mL异丁醇萃取,以每秒振摇2次的频率振摇20s,使缔合物沉淀完全溶入有机相中。立即将此异丁醇有机相吸入吸收池中,相对于参比溶液,在有色缔合物异丁醇溶液的最大吸收波长470nm处,2~15分钟内完成吸光度的测定。
2.2.2.9 回收率实验
准确吸取5.00mL丙吡胺标准溶液,移入10mL量瓶中,用注射用水稀释至刻度,摇匀。制得丙吡胺人工模拟注射溶液。分别量取丙吡胺人工模拟注射溶液0、1.00、2.00、4.00mL,至4支20mL比色管中,向各管(编号为1、2、3、4)中分别加入1.00mL丙吡胺标准溶液,加入0. 50mL醋酸-醋酸钠缓冲溶液,1.50mL四苯硼钠标准溶液,将此含丙吡胺-四苯硼沉淀的浑浊液湿过滤,分离并用去离子水充分洗涤此沉淀,将全部洗涤液放在20mL比色管中并入滤液,加入0. 50mLCu2+标准溶液、0. 50mL5%盐酸羟胺溶液,摇匀,放置1min后,加入1.00mL6.20 ×10-3mol/L的2,9-二甲基-1,10-菲罗啉溶液。用去离子水调节各管液面恰至10mL刻度线处,用1号管中溶液调吸光度为零,测定2~4号管中溶液调吸光度.。
同上操作,但不加丙吡胺标准溶液。计算回收率= [(丙吡胺标准溶液加入量与丙吡胺人工模拟注射溶液中丙吡胺的总量-丙吡胺人工模拟注射溶液中丙吡胺量)除以已知加入的丙吡胺标准溶液中的丙吡胺量]乗以 100% ,结果见表3。

表3 回收率实验
2.2.2.10 重现性
平行准确量取5.00mL丙吡胺模拟注射液3份,分为3组。按以上过程,在当天每组都重复操作两次,取平均值;然后在次日每组再重复操作一次。分别计算出各组的日内和日间相对标准偏差RSD,结果见表4。
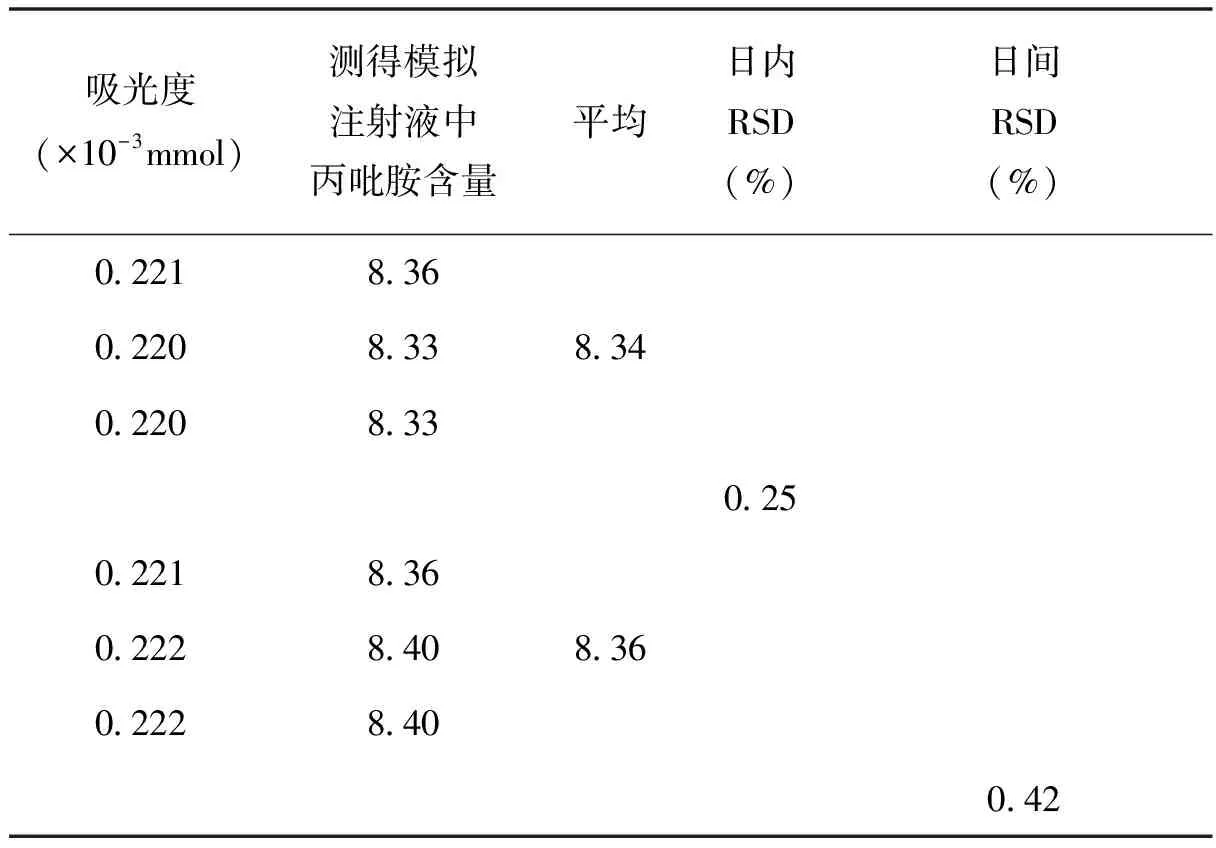
表4 本法日内和日间相对标准偏差RSD
2.2.2.11 干扰试验
在最佳条件下测定,考察外来离子存在的影响。结果表明,测定3×10-4mmol的 Ph4B-时,≦ 2×10-4mmol量的Na+,K+,Ag+,Mg2+,Zn2+,Co2+,Cd2+,Al3+及≦5×10-5mmol量的Ca2+,Sr2+,Ba2+,Fe2+,Fe3+和5×10-4mmol量的Cl-,NO3-不干扰。而5×10-5mmol量的Cu2+能抢夺Cu+-2,9-二甲基-1,10-菲罗啉螯合物中的2,9-二甲基-1,10-菲罗啉,从而有干扰,但加入盐酸羟胺将Cu2+还原成Cu+,可消除此干扰。
[1]倪文生,李安良主编.药物化学(面向21世纪课程教材)中的《心血管药物》(李长华撰写)[M]2版,北京:高等教育出版社,2000:297-299.
[2] Bortocan R, Lanchote V L,Cesarino E J, Bonato P S. Enantioselective analysis of disopyramide and mono-N-dealkyl disopyramide in plasmaand urine by high-performance liquid chromatographyon an amylose-derived chiral stationay phase[J]. J Chromatogr,B: Biomed Appl,2000,744(2):299-306.
[3]Echizen H,Ochiai K,Keto Y, Chiba K,Ishizake T.Simultaneous determination of and mono-N-dealkyl disopyramide enantiomers in plasma and urine by use of a chiral cellulose-derivative column[J]. Clin Chem, 1990,36(7):13001304.
[4] Chiba R, Yamamoto N, Tanaka A. High-performance liquid chromatography with chemiluminescene detection of disopyramide in human serum[J]. Anal Sci,1998,14(6):1153-1155.
[5] Hanada E, Ohtani H, Kotaki H, Sawada Y, Sato H, Iga T. Pharmacodynamic analysis of the electrocardiographic interaction between disopyramide and erythromycin in rats [J].J Pharm Sci, 1999, 88(2):234-240.
[6] Witek A, Znvisa T. Przyboroioski I .Determinatuon of disopyramide in plasma by High-performance liquid chromatography[J].J Pharm Biomed Anal, 1994, 12(3):425-427.
[7] Xu R. Revered-phase HPLC determination of disopyramide and its metabolite in plasma[J].Zhongguo Yiyao Gongye Zazhi, 1991,22(1):120-122.
[8] Mayer F, Kralmet B K, Ress K M, Kwhlkamp V, Liebich H M, Risler T, Seipel L. Simplified,rapid and inexpensive extraction procedure for a High-performance liquid chromatographic method for determination of disopyramide and its main metabolite mono-N-dealkyl disopyramide in serum[J]. J Chromatogr B, Biomed Appl, 1991,572(1-2):339-345.
[9] Del Cont Bernard A, Royer Morrot M J, Zhiri A, Rambourg M, Roger R J. Automated determination of disopyramide and N- mono-N-dealkyl disopyramide in plasma by reversed-phase liquid Chromatography with a colimn switching system[J]. J Chromatogr B,Biomed Appl,1992,574(2):365-368.
[10] Hanada K, Akimoto S, Hashiguchi M, Ogata H. Quantitative determination of disopyramide,verapamil and flecainide enantiomers in rat plasma and tissues by High-performance liquid chromatography[J]. J Chromatogr, B Biomed Appl, 1998, 710(1-2):129-135.
[11] Bortocan R, Lanchote L, Cesarino E J, Bonato P S. Enantioselective analysis of disopyramide and mono-N-dealkyl disopyramide in plasma and urine by High-performance liquid chromatography on an amylose-derived chiral stationary phase[J]. J Chromatogr, B Biomed Appl, 2000,744(2):299-306.
[12] Hopkala h, Drozd j, Zareba S, Disopyramide ion-selective plastic membrane sensors and their pharmaceutical applications[J].Pharmazie,1999, 54(8):600-602.
[13]Stefan R I.Lauryl surfate as counter ion for construction of ion-selective membrane electrodes for motlobemide and disopyramide[J]. Anal Chim Acta,1997,350(1-2):105-108.
[14]Amini A, Pettersson C, Westerlund D. Enantiosolution of disopyramide by capillary affinity electrokinetic chromatography wuth human1-acid glycoprotein (AGP) as chiral selector appling a partial filling technique[J]. Electrophoresis.1997,18(6): 950-957.
[15]方梅,胡波,侯媛媛,肖丹,毛细管电泳在基因研究中的应用[J],生物技术通报,2006(2): 60-66.
[16]方梅,江晓玲,胡波.21世纪毛细管电泳在食品分析中的应用[J],食品工业科技, 2006,(3):192-196,199.
[17]方梅,胡波,田贵容,徐恒,方国桢. 21世纪毛细管电泳在刑侦分析中的应用[J],分析仪器,2011(1):1-10.
[18]Kita Y, Kuroda Y, Shibukawa A, Nakagawa T.Protein binding analysis of human α1-acid glycoprotein with or without biantennary glycan[J]. Chromatography(Japanese),1999,20(4):356-357.
[19] Polisel Jabor V A, Lanchote V L, Bonato P S.Simultaneous determination of disopyramide and mono- N -dealkyl disopyramide enantiomers in human plasmaby capillary electrophoresis[J]. Electrophoresis,2001,22(7):1406-1412.
[20] Karoda Y, Matsamoto S, Shibukawa A, Nakagawa T. Capillary electrophoretic study on pH dependence of enantioselective disopyramide binding to genetic variants of humanα1-acid glycoprotein[J].Analyst, 2003,128(8):1023-1027.
[21] Fang Mei, The preliminary studies on the pharmacokinetics about the enantiomers of three kinds of cardiovascular drug. International Congress on Analytical Sciences 1-P153, June 25-30, 2006, Moscow,Russia.
[22] Fang Mei, Tian Guirong. The determination of isopyramide enantiomers by CZE. Procrrdings of The 14thNatuinal Conference on Electrochemistry, November B-018,p.198-199. 2th-6th,2007, Yangzhou,China,
[23]方梅,徐恒.测定心血管药物氯噻酮、丙吡胺的毛细管电泳研究.第十四次全国电化学会议论文集(上集),2007:200-201.
[24]高鸿,示波极谱滴定[M].南京:江苏科学技术出版社,1985:6.
[25]高鸿,示波药物滴定[M].成都:四川教育出版社,1992:49.
[26]高鸿,示波药物滴定[M].成都:四川教育出版社,1992:180.
Newspectrophotometryfordeterminationofantiarrhythmicdisopyramide.
FangMei,LongWen
(CollegeofHuaxiPharmacy,SichuanUniversity,Chengdu610041,China)
It is based on the fellowing four steps:(1)The nitrogen atoms in the disopyramide molecule reacts quantitatively with excessive sodium tetraphenylboron to form precipitate;(2)The residual anion of tetraphenylboron in filtrate forms yellow associate molecule with copperion(I)-2,9-dimethyl phenanthroline chelate in the buffer medium of acetic acid-sodium acetate;(3)The yellow associate moleculeis quantitatively extracted by isoamylol;(4)Then absorbance of the isoamylol extact layer of the yellow associated moleculeis determined in comparision with reference solution at 470 nm.. Under optimum conditions, the coefficient of linear regression is R2=0.9991,the Beer's law is obeyed in the concentration range of 0-2(×10-4)mol/L of disopyramide. The method is applicated to determinate isopyramide in the simulated inject solution and the recoveries of standard addition are 99.76%-100.66%.Within- and between-day coefficients of variation are 0.25% and 0.42%, respectively. When determing 3×10-4mmol Ph4B-,under the conditions of ≤2×10-4mmol of Na+,K+,Ag+,Mg2+,Zn2+,Co2+,Cd2+,Al3+,≤5×10-5mmol of Ca2+,Sr2+,Ba2+,Fe2+,Fe3+and ≤5×10-4mmol of Cl-,NO3-does not interference with the results. The method is simple and reliable.
antiarrhythmic;disopyramide(mixture of enantiomers); spectrophotometry;sodium tetraphenyl boron; 2,9-dimethyl-1,10-phenathroline;extraction
国家自然科学基金资助项目,项目编号:20475038。
10.3969/j.issn.1001-232x.2017.06.017
2017-09-21

方梅,四川大学化学系学士,硕士,中国科学院北京化学研究所博士,中国科学院大连化学物理研究所博士后,美国印第安纳大学伯明翰分校化学系高级研究学者。先后任华西医科大学药学院助教、讲师,四川大学华西药学院副研究员,招收三名硕士生,指导多名本科毕业生和进修教师学术论文,E⁃mail:abc843@163com。
猜你喜欢
中老年保健(2022年6期)2022-08-19
中国油脂(2019年10期)2019-11-20
现代食品(2018年13期)2018-02-14
Coco薇(2017年8期)2017-08-03
广东饲料(2016年2期)2016-12-01
现代农业(2015年3期)2015-02-28
现代畜牧科技(2015年7期)2015-02-23
应用化工(2014年1期)2014-08-16
中国有色金属(2014年2期)2014-04-11
语文教学与研究(2014年11期)2014-02-28
