
抗体药物偶联物的弹头分子研究进展
2017-12-13 02:13胡馨月李艳萍李卓荣
中国医药生物技术 2017年6期
胡馨月,李艳萍,李卓荣
抗体药物偶联物的弹头分子研究进展
胡馨月,李艳萍,李卓荣
在肿瘤治疗中,化疗为主要的治疗手段,在杀死肿瘤细胞的同时,也对正常细胞存在杀伤作用,从而产生严重的毒副作用。如何将细胞毒性药物准确输送到病灶部位,选择性地杀伤肿瘤细胞,渐渐引起了人们的重视。追溯到 100 年前,Paul Ehrlich 就提出了利用抗体靶向治疗癌症的“魔术子弹”概念[1]。随着抗体技术的发展,掀起了抗体药物偶联物(antibody-drug conjugate,ADC)的研究浪潮,ADC 是抗体与细胞毒药物的完美“联姻”。ADC 是由“抗体”、“连接子”和“弹头分子”三个主要组件构成,抗体药物偶联物能够特异性识别肿瘤抗原,形成的 ADC-抗原复合物通过受体介导的胞吞作用进入靶细胞内部,释放出高活性的弹头分子,完成对肿瘤细胞的选择性杀伤(图 1)[2]。

图 1 抗体药物偶联物的作用机制
理论上说化疗药物、细胞毒素、放射性核素等对肿瘤细胞具有较大杀伤作用的细胞毒性物质都可以作为 ADC 的弹头分子,通常必须同时具备以下三点特征:①作用机制明确。基于 ADC 的作用机制,理想的弹头分子的靶点应位于细胞内,如抗有丝分裂或者抑制 DNA 复制。②弹头分子的细胞毒作用必须极高,一般要求 EC50小于 1 nmol/L。研究表明放射标记的抗体在癌症病人肿瘤部位摄取量很低(每克肿瘤组织仅仅集中 0.003% ~ 0.01% 的抗体注射剂量)[3],所以弹头分子需在较低浓度下也可发挥作用。③可以被修饰,允许被连接。天然发现的细胞毒药物往往没有可以连接的位点,大多数需经过结构改造,使之成为具有可连接的基团且与天然结构生物活性相当的衍生物。
为降低传统的化疗药物(如甲氨蝶呤、环磷酰胺、长春碱类、紫杉烷类)毒副作用,增加它们的靶向性,在设计第一代抗体药物偶联物时,人们曾尝试将它们偶联在抗体上。之后很快发现,这类 ADC 在临床使用中无法达到预期的抗肿瘤活性,甚至低于化疗药物本身的效能[4],其主要原因在于 ADC 经注射后富集于靶部位的剂量低,加之化疗药物本身细胞毒性不够强,治疗效果大大减弱。随之研究人员在植物、海洋生物和微生物等天然来源的化合物中寻找新型高效细胞毒性化合物,研究发现某些化合物在体外对多种肿瘤细胞的抑制活性比传统化疗药物高 100 ~ 1000 倍[5],IC50值能够达到 pmol/L 水平。这些细胞毒素由于治疗安全窗范围较窄,不能作为药物单独使用,故可以考虑用来做 ADC的弹头分子发挥作用。本文将对目前临床在研 ADC 弹头分子的进展进行综述。
1 微管蛋白抑制剂
1.1 海兔毒素及奥瑞他汀类
海兔毒素(dolastatin)是从印度洋生物截尾海兔中发现的一类具有高细胞毒性的线性多肽类化合物。Pettit 等[6]表征了海兔毒素中多种毒性组分,并且阐明了海兔毒素 10 的作用机制和结构(图 2,1),这类细胞毒药物能够显著抑制微管蛋白的形成和聚合。海兔毒素 10 对白血病细胞L1210(IC50= 0.5 nmol/L)等肿瘤细胞具有普遍杀伤能力,对细胞的抑制活性约为长春碱的 40 倍,根霉素的 2 倍,拟茎点霉毒素 A 的 10 000 余倍[7],然而海兔毒素 10 在单独使用时存在毒副作用大、药代动力学性质差、治疗窗口窄等问题限制了其开发。Miyazaki 等[8]发现 N 端为仲胺的单甲基奥瑞他汀 D(monomethyl auristatin D,MMAD)毒性与海兔毒素 10 相当,将这类 N-单甲基取代的海兔毒素衍生物命名为奥瑞他汀类(auristatin),它们可实现与连接子的有效连接,新一代的高效 ADC 也由此产生[9]。

图 2 海兔毒素类细胞毒药物和 MMAE、MMAF 类抗体药物偶联物
目前,ADC 研发常用的弹头分子为奥瑞他汀类化合物MMAE 和 MMAF(图 2,2 和 3)。与 MMAD 相比,MMAE 的 C 端为 2-氨基-1-苯基丙基-1-醇,IC50值为10-11~ 10-9mol/L。由 Seattle Genetics 公司研发的治疗霍奇金淋巴瘤和系统型间变性大细胞淋巴瘤的 Adcetris(SGN-35)在 2011 年已被 FDA 批准上市,该药为抗CD30 人鼠嵌合抗体,采用缬氨酸-瓜氨酸(Val-Cit)酶解型连接子与 MMAE 连接,通过还原抗体链间 4 对二硫键,连接 MMAE 个数为 0、2、4、6、8 不等。研究发现,平均药物/抗体偶联比以 4 为最佳,该 ADC(图 2,4)在细胞中可以完全释放出细胞毒药物 MMAE[10]。II 期临床试验报道,102 例患者以 1.8 mg/kg 给药三周,霍奇金淋巴瘤病人总缓解率为 75%(34% 全响应,40% 部分响应),系统型间变性大细胞淋巴瘤病人总缓解率为 87%(53% 全响应),治疗效果显著[11]。目前,至少 4 项作为一线用药的 III期临床试验仍在进行,以期进一步扩大其适应证范围。MMAF 的 C 端为苯丙氨酸,体外细胞毒性比 MMAE 弱100 倍[12],主要由于本身羧基的离子化,使得 MMAF 的膜通透性较差,阻碍了它扩散进入细胞。以 MMAF 为弹头分子的 ADC(图 2,5)通常使用不可裂解型连接子马来酰亚胺基己酸(MC),在细胞内最终降解为带有半胱氨酸(Cys)残基的 MC-MMAF 形式,仍能够保持很好的活性。目前,临床在研 ADC 约 70 多个,以 MMAE 和 MMAF作为弹头分子的 ADC 占有主导地位(表 1)。
1.2 美登素及美登素类衍生物
美登素(maytansine)是 Kupchan 等[13]于 1972 年在非洲灌木丛美登木中提取分离出来的。美登素(图 3,6)对人口腔表皮样癌细胞 KB 的 ED50为 10-4~ 10-5μg/ml,效果可达长春花碱的 100 ~ 1000 倍,具有良好的稳定性和溶解性。由于天然的美登素结构中没有连接基团,经研究者对美登素的构效关系深入探究之后,C3 位可被改造为末端有巯基的新型酯类连接位点[14]。DM1 和 DM4(图 3,7 和8)正是对 C3 位修饰后的一类美登素衍生物,此类化合物是由发酵得来的安丝菌素还原成美登醇,C3 位与连有二硫键的酸缩合,最终还原成末端巯基得到。

表 1 批准上市和临床在研的奥瑞他汀类 ADC

图 3 美登素类细胞毒药物和 DM1、DM4 类抗体药物偶联物
以美登素类作为弹头分子的 ADC 一般采用与抗体赖氨酸残基偶联的方式,DM1 型 ADC(图 3,9)通常使用不可裂解型双功能连接子 SMCC(N-succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate),在细胞内经溶酶体降解为 Lys-SMCC-DM1 形式。DM4 型 ADC(图3,10)一般使用可裂解型连接子为 SPDB(N-succinimidyl 4-(2-pyridyldithio) butyrate) 或 SPP( N-succinimidyl 4-(2-pyridyldithio) pentanoate),在细胞内首先分解为Lys-SPDB/SPP-DM4,再经内源性谷胱甘肽还原,二硫键断裂成 SH-DM4,直接发挥作用或部分甲基化发挥更强的抗肿瘤作用。Kadcyla(T-DM1)即曲妥珠单抗-SMCC-DM1,在 2013 年批准治疗转移性晚期乳腺癌,对于 HER-2 阳性晚期乳腺癌病人使用 T-DM1 组较曲妥珠单抗与紫杉烷组毒副作用显著降低,针对病人无进展生存时间和总生存时间均优于拉帕替尼与卡培他滨联合用药组[15]。另外,该药用于晚期胃癌、非小细胞肺癌等疾病的治疗也进入临床研究中。以美登素类细胞毒药物为弹头分子进入临床评价的ADC 如表 2 所示。

表 2 临床在研和已上市美登素类抗体药物偶联物

图 4 卡奇霉素类和卡奇霉素类抗体药物偶联物
2 DNA 损伤剂
2.1 卡奇霉素类
卡奇霉素(calicheamicin)(图 4,11)是一类作用于DNA 的抗肿瘤抗生素,1986 年从棘孢小单孢菌中得到的发酵产物[16],属于典型的烯二炔类化合物,结构中含有一个 1,5-二炔-3-烯母核,一个全取代的苯,四个糖苷和一个特殊的三硫键。卡奇霉素通过低聚糖片段与 DNA 结合,嵌入 DNA 双螺旋小沟的 5'-TCCT-3' 和 5'-TTTT-3' 位点,烯二炔结构通过 Bergman 环化发生电子重排,关环成苯环双自由基,夺取 DNA 链上的 H 原子,导致 DNA 链断裂。其中,研究发现卡奇霉素 γ1I对体内多种肿瘤有着极好的效能,使用剂量最低达到 0.15 μg/kg[17]。基于卡奇霉素 γ1I良好的抗肿瘤特性,设计了一种卡奇霉素衍生物 N-乙酰基-γ-卡奇霉素,天然的三硫键结构改造为二硫键,可实现与腙键型或酰胺型的连接子的连接[18]。FDA 批准上市的第一个ADC 药物 Mylotarg 就是通过腙键型连接子将抗 CD33的吉妥珠单抗与 N-乙酰基-γ-卡奇霉素-DMH 偶联(图 4,12a),2009 年被批准治疗急性髓系白血病。临床使用中,该药在 9 mg/m2的剂量下,大约有 30% 的患者得到完全缓解,但随后发现它存在连接子不稳定,抗体 FAB 段与血清抗体易发生交换等问题,在治疗过程中部分病人出现致命性肝损伤,迫使其于 2010 年撤市。
目前值得关注的是 CMC-544(图 4,12b),现已发现其在体外有特异性好、稳定性强及活性高等优点,在重症联合免疫缺陷小鼠多种淋巴瘤模型中,该 ADC 可以持续抑制肿瘤生长近 100 d[19],针对于急性淋巴细胞白血病的治疗已进入 III 期临床[20]。PF-006647263(图 4,12c)是以 ephrin A4 为靶点,在化学、生产和控制方面均进行了合理优化,得到平均 DAR 为 4 且没有裸抗体存在的 ADC[21],I 期临床用于三阴型乳腺癌的治疗。
2.2 倍癌霉素类
倍癌霉素 A(图 5,13)和倍癌霉素 SA(图 5,14)是 19 世纪 70 年代从链霉菌属 zelensis 细菌培养物中分离得到[22],该类化合物能够嵌入 DNA 小沟中 A-T 结构域丰富的位置,经腺嘌呤的 N3 位进攻其环丙基基团,造成DNA 烷基化,导致细胞死亡,具有抗菌、抗肿瘤功能。MDX-1203(图 5,15)使用的倍癌霉素类似物 MED-2460是将 CBI 结构单元(图 5,16)苯酚改造为甲基哌嗪甲酸酯前药形式,环丙基开环替换成氯甲基,吡咯上的 N 连接氨基取代吲哚。该 ADC 靶向于 CD70 抗原,DAR 为1.25,在肾细胞癌 786-O 裸鼠模型中,以 38 mg/kg 剂量给药两次后肿瘤可完全消退,I 期临床试验用于治疗肾上皮细胞癌和非霍奇金淋巴瘤,药物安全性高,病人以 8 mg/kg的剂量耐受性良好[23]。SYD985(图 5,17)选用的弹头分子 是 倍 癌 霉 素 咪 唑 并 [1,2-α]吡 啶 系 列 衍 生 物seco-DUBA[24],通过还原曲妥珠单抗链间二硫键偶联,经疏水作用层析分离后使得每个抗体平均连接 2.8 个seco-DUBA[25],该 ADC 也已进入 I 期临床,治疗多种非特异性实体瘤,主要是以 HER2 过表达的实体瘤为主。

图 5 CBI 单元、倍癌霉素类和倍癌霉素类抗体药物偶联物
2.3 安曲霉素类衍生物 PBD
19 世纪 60 年代,从链霉菌属 Streptomyces refuineus var. thermotolerans 中发酵提取分离得到安曲霉素[26](图 6,18),并发现了一类具有抗菌、抗肿瘤的结构类似物 PBD(pyrrolobenzodiazepine,吡咯并苯并二氮杂䓬)家族分子,研究共发现了 13 种不同 PBD 天然单体类型,它们均是由吡咯环 A、二氮杂䓬 B 和芳环 C 组成,通过 N10/C11 亚胺功能团与 DNA 小沟鸟嘌呤的 C2 位氨基作用而发挥生物学功能。随后,有人进一步探究发现 PBD 二聚体较 PBD与 DNA 作用面积更大,可与鸟嘌呤形成两个共价键,增加了序列的选择性[27],且二聚体对于多种肿瘤细胞系(如乳腺癌细胞 MCF-7、卵巢癌细胞 A2780 等)的活性达到pmol/L 级别,如 DSB-120(图 6,19)是由两分子 DC-81(图 6,20)通过丙二醚键相连,以 GATC 序列为中心横跨了 6 个碱基对与 DNA 交联,相对于 DC-81 单体体外活性增加了 600 倍。自 2013 年以来,PBD 二聚体这种高效作用于 DNA 的特性逐渐作为新型 ADC 弹头分子被使用,地位仅次于海兔毒素类和美登素类。Seattle Genetics 公司研发的一系列以 PBD 为弹头分子的 ADC 都是以酶裂解性缬氨酸-丙氨酸(VA)为连接子,通过工程化半胱氨酸的偶联技术得到平均 DAR 为 1.9。目前有 5 个靶点的ADC(图 6,21):SGN-CD33A、SGN-CD70A、SGN-CD19B、SGN-CD123A、SGN-352A 分别治疗急性髓系白血病(III期)[28]、肾上皮细胞癌(II 期)[29]、非霍奇金淋巴瘤(I 期)、急性髓系白血病(I 期)和多发性骨髓瘤(I 期)。
2.4 喜树碱类衍生物 SN-38
喜树碱(图 7,22)通过作用于 DNA 拓扑异构酶 Ӏ 抑制 DNA 的复制和转录,导致肿瘤细胞死亡,具有强体外抗肿瘤活性。然而,喜树碱水溶性差,内酯环易开环,从而降低了生物利用度,限制其临床应用。设计带有哌嗪环前药的喜树碱类衍生物伊立替康(图 7,23),它在体内以关闭的内酯和开环的羧酸盐两种形式存在,代谢产物 SN-38(图 7,24)为主要的抗肿瘤成分。SN-38 的 C20 位羟基除了能够降低内酯环在体内降解速率外,还可以与连接子相连用于 ADC 的开发。Labetuzumab-SN-38(IMMU-130,图 7,25a)为抗 CD66-CL2(赖氨酸-苯丙氨酸)-SN-38,临床前研究显示其可显著延长直肠癌和胰腺癌小鼠生存率,目前 II 期临床治疗复发/难治型结肠癌。IMMU-132(图7,25b,抗 Trop-2-SN-38)是通过 CL2A(赖氨酸,Lys)型连接子连接人源化单抗 hRS7 和 SN-38,由于 Trop-2 抗原能够广泛表达在肿瘤滋养层细胞表面[30],因此 IMMU-132在大肠癌、食管癌、三阴性乳腺癌、小细胞肺癌和非小细胞肺癌患者中均取得了部分响应。

图 6 安曲霉素、PBD 类和 PBD 类抗体药物偶联物
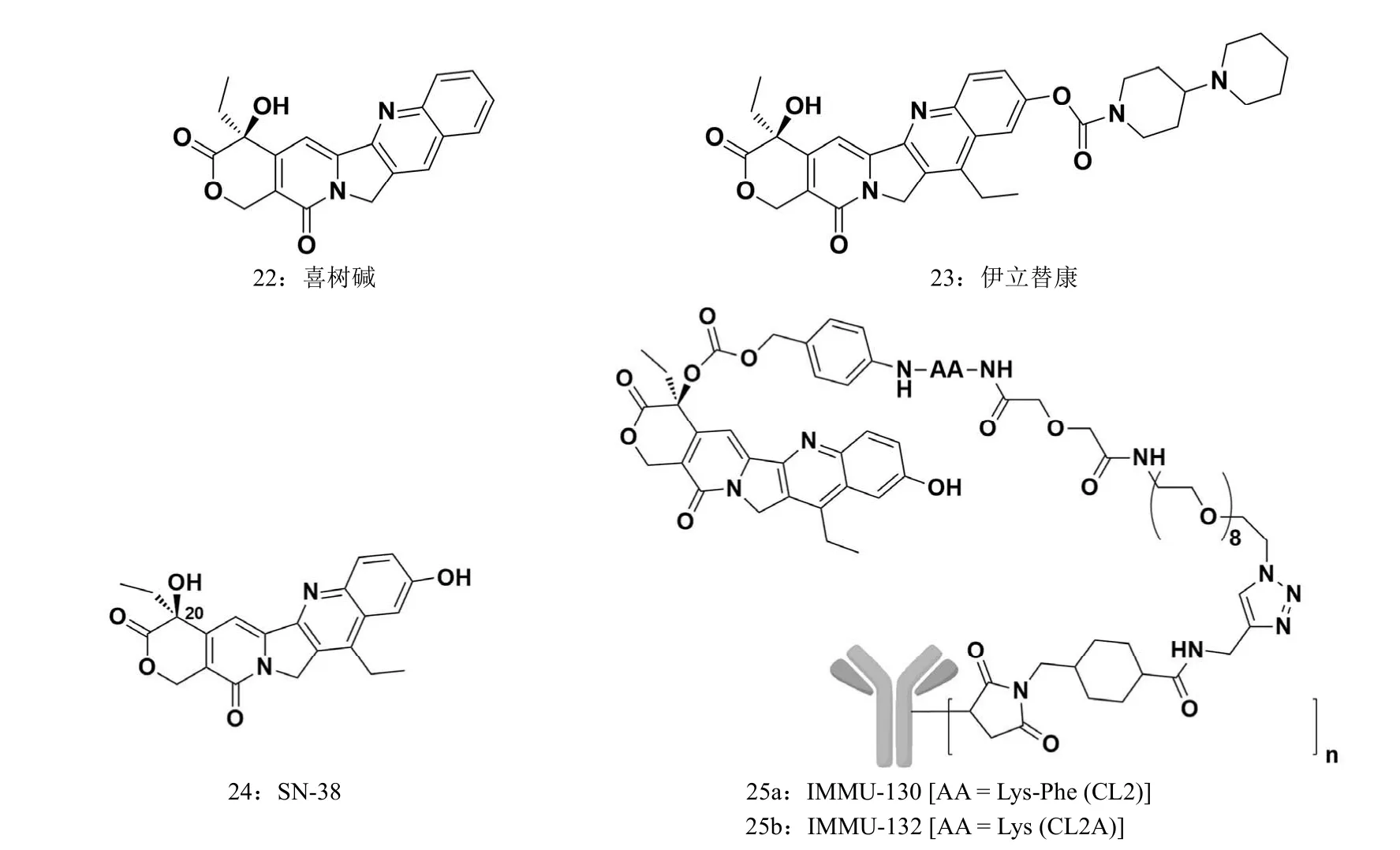
图 7 喜树碱类和 SN-38 类抗体药物偶联物
3 结语
ADC 解决了细胞毒药物选择性差的问题,使得某些细胞毒性过强的药物有了用武之地,同时也为肿瘤的靶向治疗提供了更多的选择和手段。寻找合适的新型 ADC 弹头分子,从细胞毒药物的发现及其作用机制的探究,再到结构改造和构效关系研究,这一过程具有巨大挑战性。而且,采用何种连接技术以及相应的连接子也是解决这些问题的关键,抗体除了半胱氨酸和赖氨酸偶联的方法外,为降低产品的异质性问题,引入非天然氨基酸、工程化半胱氨酸和定点突变等技术均有报道[31-32];连接子的优化着重在于增加水溶性和稳定性,如能够被 β-葡萄糖醛酸水解的葡萄糖醛酸型连接子[33],降低细胞毒药物脱靶的双功能二硫桥连接子[34]等将为理想 ADC 的设计提供有效策略。总之,随着新技术和新策略的不断涌现,ADC 在肿瘤甚至其他疾病治疗中的应用将会蓬勃发展。
[1] Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer, 2008, 8(6):473-480.
[2] Gordon MR, Canakci M, Li L, et al. Field guide to challenges and opportunities in antibody-drug conjugates for chemists. Bioconju Chem, 2015, 26(11):2198-2215.
[3] Sedlacek HH, Seemann G, HoffmannD, et al. Antibodies as carriers of cytotoxicity. Contrib Oncol Basel Karger, 1992, 43:1-45.
[4] Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol, 2009, 13(3):235-244.
[5] Pietersz GA, Rowland A, Smyth MJ, et al. Chemoimmunoconjugates for the treatment of cancer. Adv Immunol, 1994, 56:301-387.
[6] Pettit GR, Srirangam JK, Singh SB, et al. Dolastatins 24: synthesis of (-)-dolastatin 10. X-Ray molecular structure of N,N-dimethylvalylvalyl-dolaisoleuine tert-butyl ester. J Chem Soc Perkin Trans 1, 1996, 8:859-863.
[7] Bai R, Pettit GR, Hamel E, et al. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem Pharmacol, 1990, 39(12):1941-1949.
[8] Miyazaki K, Kobayashi M, Natsume, T, et al. Synthesis and antitumor activity of novel dolastatin 10 analogs. Chem Pharm Bull (Tokyo), 1995, 43(10):1706-1718.
[9] Perez HL, Cardarelli PM, Deshpande S, et al. Antibody-drug conjugates: current status and future directions. Drug Discov Today, 2014, 19(7):869-881.
[10] Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res, 2004, 10(20):7063-7070.
[11] Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med, 2010, 363(19):1812-1821.
[12] Doronina SO, Mendelsohn BA, Bovee TD, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery:effects of linker technology on efficacy and toxicity. Bioconju Chem, 2006, 17(1):114-124.
[13] Kupchan SM, Komoda Y, Court WA, et al. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J Am Chem Soc, 1972, 94(4):1354-1356.
[14] Cassady JM, Chan KK, Floss HG, et al. Recent developments in the maytansinoid antitumor agents. Chem Pharm Bull (Tokyo), 2004, 52(1):1-26.
[15] Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med, 2012, 367(19):1783-1791.
[16] Smith AL, Nicolaou KC. The enediyne antibiotics. J Med Chem, 1996, 39(11):2103-2117.
[17] Lee AD, Dunne TS, Siegel MM, et al. Calichemicins, a novel family of antitumor antibiotics. 1. Chemistry and partial structure of calichemicin gamma.1I. J Am Chem Soc, 1987, 109(11):3464-3466.
[18] Hamann PR, Hinman LM, Beyer CF, et al. An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconju Chem, 2002, 13(1):40-46.
[19] DiJoseph JF, Dougher MM, Evans DY, et al. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother Pharmacol, 2011, 67(4):741-749.
[20] Spiluttini B, Gu B, Belagal P, et al. Splicing-independent recruitment of U1 snRNP to a transcription unit in living cells. J Cell Sci, 2010, 123(12):2085-2093.
[21] Damelin M, Bankovich A, Park A, et al. Anti-EFNA4 calicheamicin conjugates effectively target triple-negative breast and ovarian tumor-initiating cells to result in sustained tumor regressions. Clin Cancer Res, 2015, 21(18):4165-4173.
[22] Hanka LJ, Dietz A, Gerpheide SA, et al. CC-1065 (NSC-298223), a new antitumor antibiotic. Production, in vitro biological activity, microbiological assays and taxonomy of the producing microorganism. J Antibiot (Tokyo), 1978, 31(12):1211-1217.
[23] Owonikoko TK, Hussain A, Stadler WM, et al. First-in-human multicenter phase I study of BMS-936561 (MDX-1203), an antibody-drug conjugate targeting CD70. Cancer Chemother Pharmacol, 2016, 77(1):155-162.
[24] Elgersma RC, Coumans RG, Huijbregts T, et al. Synthesis, and evaluation of linker-duocarmycin payloads: toward selection of her2-targeting antibody-drug conjugate SYD985. Mol Pharm, 2015, 12(6):1813-1835.
[25] Black J, Menderes G, Bellone S, et al. SYD985, a novel duocarmycin-based HER2-targeting antibody-drug conjugate, shows antitumor activity in uterine serous carcinoma with HER2/neu expression. Mol Cancer Ther, 2016, 15(8):1900-1909.
[26] Leimgruber W, Stefanović V, Schenker F, et al. Isolation and characterization of anthramycin, a new antitumor antibiotic. J Am Chem Soc, 1965, 87(24):5791-5793.
[27] Bose DS, Thompson AS, Ching JS, et al. Rational design of a highly efficient irreversible DNA interstrand cross-linking agent based on the pyrrolobenzodiazepine ring system. J Am Chem Soc, 1992, 114(12):4939-4941.
[28] Kung Sutherland MS, Walter RB, Jeffrey SC, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood, 2013, 122(8):1455-1463.
[29] Jeffrey SC, Burke PJ, Lyon RP, et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconju Chem, 2013, 24(7):1256-1263.
[30] Cardillo TM, Govindan SV, Sharkey RM, et al. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin Cancer Res, 2011, 17(10):3157-3169.
[31] Behrens CR, Liu B. Methods for site-specific drug conjugation to antibodies. MAbs, 2014, 6(1):46-53.
[32] Yao H, Jiang F, Lu A, et al. Methods to design and synthesize antibody-drug conjugates (ADCs). Int J Mol Sci, 2016, 17(2):pii:E194.
[33] Lu J, Jiang F, Lu A, et al. Linkers having a crucial role in antibody-drug conjugates. Int J Mol Sci, 2016, 17(4):561.
[34] Badescu G, Bryant P, Bird M, et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconju Chem, 2014, 25(6):1124-1136.
10.3969/j.issn.1673-713X.2017.06.010
中国医学科学院医学与健康科技创新工程(2016-I2M-3-022);北京协和医学院协和青年科研基金(2017350021)
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所
李卓荣,Email:l-z-r@263.net
2017-09-04
猜你喜欢
兵工学报(2022年9期)2022-10-11
作物学报(2022年7期)2022-05-12
化学研究(2021年4期)2021-08-19
世界最新医学信息文摘(2021年12期)2021-06-09
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
中国药科大学学报(2020年1期)2020-05-12
四川蚕业(2020年4期)2020-02-10
现代防御技术(2018年5期)2018-10-29
现代防御技术(2014年6期)2014-02-28
恋爱婚姻家庭·养生版(2013年6期)2013-05-14
