
陆地棉MIKCC基因家族的全基因组分析
2017-12-04 08:00周娜汪露瑶张天真胡艳
棉花学报 2017年6期
周娜,汪露瑶,张天真,胡艳
(南京农业大学作物遗传与种质创新国家重点实验室,南京210095)
陆地棉MIKCC基因家族的全基因组分析
周娜,汪露瑶,张天真,胡艳*
(南京农业大学作物遗传与种质创新国家重点实验室,南京210095)
【目的】MIKCC是1类保守的转录因子家族,参与调控植物的开花时间和花器官发育。通过对MIKCC家族进行全基因组学分析,为深入研究MIKCC在棉花开花及花器官发育的分子调控机理提供基础。【方法】利用HMMER 3.0及pfam种子文件鉴定棉花全基因组MIKCC基因,结合其表达量进行偏向表达及聚类分析。【结果】在陆地棉基因组中共鉴定发现100个MIKCC基因,分为11个亚类。表达聚类分析显示,棉花MIKCC基因的表达模式大致可分为4个不同的类群,说明这类基因在棉花进化中出现了功能分化。100个MIKCC基因中,有12个基因具有miRNA靶位点。【结论】研究结果显示MIKCC家族基因在棉花纤维中存在功能分化,而且可能受到miRNA的调控,这为进一步研究该家族基因的功能提供信息参考。
陆地棉;MIKCC基因;功能分化;miRNA
MIKCC是1类具有MADS和K-box结构域的植物特异转录因子,属于MADS-box蛋白家族。研究发现MADS-box转录因子在调控植株的生长发育,特别是在花器官形成中发挥重要作用[1-2]。基于聚类分析的结果,MADS-box被分为I型和II型2个大类[1-2]。I型MADS-box基因通常含有1~2个外显子,但是它并不含Kertain-like结构域(又称K-box或K结构域),有时也称其为M型基因[3]。II型的MADS-box基因也叫MIKC型,具有多内含子和多外显子结构。典型的MIKC类蛋白含有4个结构域,分别是MEF2-like结构域 (M 结构域)、Intervening domain (I结构域)、Kertain-like结构域和C端结构域 (C结构域)[4]。MIKC型又被分为MIKCC类型和MIKC*类型。MIKCC基因和MIKC*基因的主要区别在于MIKC*类的I结构域更长且没有K结构域[5-6]。目前在拟南芥中,总共发现7个MIKC*基因和39个MIKCC基因,这39个基因依据功能不同分为12 个亚类[7]。
近来的研究表明,MIKCC在植物发育中发挥非常重要的功能,参与花器官的发育、控制开花时间[8],参与营养器官的发育[9]、种皮着色和胚胎发育[10]等。MIKCC蛋白在控制开花时间和花器官发育方面的作用尤为重要。传统上认为,花器官发育由 A(APETALA1,AP1),B(PISTILATA,PI和APETALA3,AP3),C(AGAMOUS,AG)3 类基因控制,而最近的研究表明,花器官发育由A,B,C和 D(SEEDSTICK,STK),E(SEPALLATA,包括SEP1,SEP2,SEP3 和SEP4)[12]共 5 类基因协同调控。不同的花器官发育被不同组合的基因控制:如萼片(A+E),花瓣(A+B+E),雄蕊(B+C+E),雌蕊(C+E)和胚珠(D+E)[11]。 近几十年来,一些关键的MIKCC基因被发现参与拟南芥开花时间的调控[13-15]。FLOWERING LOCUS C(FLC)基因抑制开花[16]。SHORT VEGETATIVE PHASE(SVP)通过温度控制开花时间[17]。AGAMOUS-LIKE16(AGL16)被 miR824 靶向,延迟植物开花[18]。与MIKCC类基因相比,对MIKC*类基因的研究相对较少。
棉纤维是纺织业最常用的天然材料。随着耕地面积减少,粮棉争地矛盾日益凸显。早熟棉种的选育推广可以缓解这一矛盾。棉花的早熟性状与多种农艺性状相关,如:苗期、花期、铃期、产量等[19]。其中花期调控是缩短棉花生育期,促进早熟的重要途径。研究发现,MIKC基因在棉花的生长发育中也发挥非常重要的作用,如GhMADS1在花瓣中的表达量最高,在营养器官中的表达水平较低,推测其调控棉花花器官的形成[20]。Gh-MADS3与拟南芥中的AG基因同源,在雄蕊和心皮中的表达水平高,主要调控雄蕊和心皮的发育[21]。GhMADS11在棉花的纤维细胞中高表达,促进纤维细胞伸长[22]。GhAGL15s在体细胞胚胎发育时期优势表达,促进棉花胚性愈伤组织的形成[23]。GhMADS22促进开花和延迟衰老[24]。随着基因组测序技术的发展,陆地棉(Gossypium hir-sutum)[25]、亚洲棉(G.arboreumL.)[26]和雷蒙德氏棉(G.raimondiiUlbr.)[27]均完成了基因组测序工作。本研究通过对陆地棉TM-1基因组数据进行结构域搜索,鉴定MIKCC基因,从基因组水平分析MIKCC基因的数目、染色体分布以及进化关系和表达模式,为解析棉花MIKC功能奠定基础。
1 材料与方法
1.1 陆地棉中的MIKCC家族基因的鉴定
陆地棉TM-1的数据来自本校Cotton Research Institute (http://mascotton.njau.edu.cn)。MIKCC基因的种子文件 SRF-TF (PF00319)和K-box(PF01486) 来自 pfam 网站 (http://pfam.xfam.org/)。 利用 HMMER3.0[28]软件对 MIKC 蛋白序列进行鉴定,HMMER运行参数-E 0.01。
1.2 MIKCC家族基因的染色体定位、进化树构建及结构分析
将棉花的MIKCC基因与已经分类的拟南芥的MIKCC基因进行生物信息分析,利用MUSCLE[29]进行序列比对,利用MEGA 5.0[30]进行系统进化树分析,使用邻近法,抽样次数1000次。利用BLASTN[31]将MIKCC核苷酸序列比对到陆地棉表达序列标签 (Expressed sequence tags,EST)数据库(参数:E 值 1 ×10-10)。 利用 Perl脚本解析棉花GFF注释文件,获取MIKCC基因家族成员的物理位置和基因结构。染色体定位软件选用Mapinspect(http://mapinspect.software.informer.com),基因结构分析结果用Gene Structure Display Server(http://gsds.cbi.pku.edu.cn/index.php) 进 行可视化。序列基序使用MEME(http://meme.sdsc.edu/meme/meme.html)进行分析,参数设置为基序长度最长100个氨基酸,基序最大发现数目4个。
1.3 MIKCC家族基因表达分析以及miRNA靶基因的预测
陆地棉的转录组数据下载自SRA数据库(http://mascotton.njau.edu.cn)中的棉花基因组测序项目(PRJNA248163),用 SolexQA[32]对数据进行质量控制,Tophat2[33]进行比对,cufflink[33]计算FPKM值作为基因表达量,pheatmap(https://cran.r-project.org/web/packages/pheatmap/index.html)进行可视化。用psRNAtarget网站(http://plantgrn.noble.org/psRNATarget/)[34]预测 MIKCC家族基因的miRNA靶基因。
2 结果与分析
2.1 全基因组鉴定陆地棉MIKCC基因家族以及进化分析
利用陆地棉TM-1蛋白和基因组序列,去冗余后,预测是否有完整的结构域,在陆地棉中鉴定出100个MIKCC基因,除了蛋白数据库注释的96个,在基因组中共找到4个新的MIKCC基因。在100个MIKCC中,共鉴定到49对A和D亚组部分同源基因对(图1),未能找到GhMADS46D和GhMADS47D的部分同源基因,推测其同源基因可能在进化过程中丢失。利用Mapinspect对MIKCC基因进行染色体定位分析。由图1可见,MIKCC基因在陆地棉26条染色体上均有分布,49个MIKCC基因定位于A亚组,50个位于D亚组,1个位于未能锚定到染色体的scaffold片段。为了阐明基因家族扩增原因,使用MCScanX[35]分析串联重复事件。结果表明,仅GhMADS31D和GhMADS43D基因经历了串联重复复制,与拟南芥的AGAMOUS(AT4G18960)为同源基因,后者与花器官雄蕊及心皮的发育相关。MIKCC家族大部分基因未发生基因的串联复制,表明串联重复复制对棉花MIKCC家族扩增贡献很小。
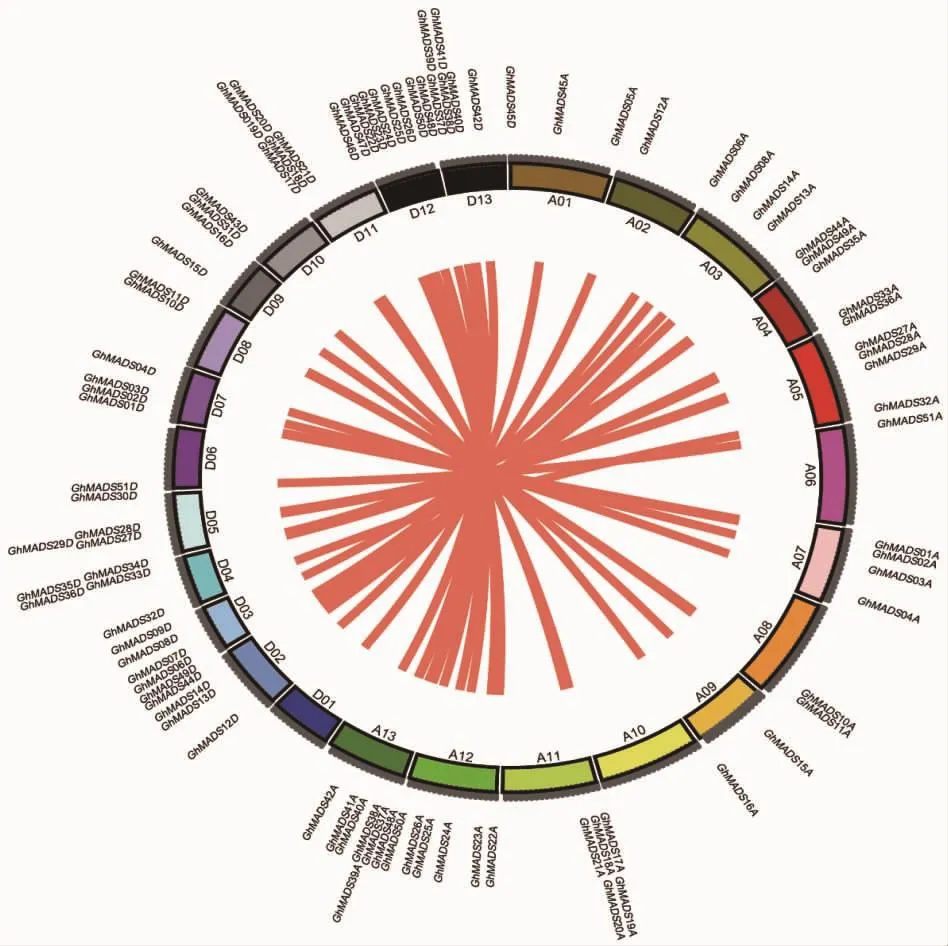
图1 陆地棉共线性部分同源MIKCC基因Fig.1 Distribution of syntenicMIKCCgenes on the 26 chromosomes ofG.hirsutum
为了确定棉花MIKCC与其他已知物种MIKCC进化关系,利用分别来自陆地棉、葡萄(Vitis vinifera)和拟南芥(Arabidopsis thaliana)的39、32和39的MIKCC基因,采用临近法构建系统发育树。再根据葡萄和拟南芥的MIKCC基因对棉花MIKCC基因进行分类。根据聚类结果,MIKCC基 因 被 分 为 A,B,C/D,E,BS,TM8,ANR1-like,AGL15AVP-like,SOC1-like,FLC-like共11类(图2)。陆地棉的MIKCC基因分布在除FLC-like亚类外的10个亚类中。

图2 陆地棉、葡萄和拟南芥基因组中MIKCC基因的系统发育分析Fig.2 Phylogenetic tree ofMIKCCgenes inG.hirsutum,Vitis viniferaandArabidopsis thaliana
2.2 陆地棉MIKCC基因的结构分析
为了解MIKCC基因结构,根据其系统发育关系,比较了MIKCC基因的内含子/外显子和保守基序。分析发现,陆地棉中的MIKCC基因长度变化很大,在5~30 kb之间,并且都有多个外显子和多个内含子(图3),与拟南芥和水稻报道的MIKCC结构一致。陆地棉有些A和D亚组部分同源基因之间结构差异大,大部分的A亚组MIKCC基因的内含子比D亚组MIKCC基因内含子长,从而导致A亚组的基因比D亚组基因长。但GhMADS44的D亚组基因比A亚组长,可能与陆地棉在进化过程中的染色体片段交换有关。
MIKCC蛋白含有4个保守区域,利用MEME对棉花MIKCC蛋白的氨基酸序列进行保守性分析。图3显示,MIKCC蛋白结构域高度保守,绝大多数的MIKCC蛋白包含4个保守的基序。通过序列分析,大部分MIKCC蛋白均含有M、I、K、C共4个结构域,有些蛋白仅有3个结构域,如陆地棉中参与配子体发育的AGL15类蛋白几乎都没有K-box结构域,这可能与此类蛋白的特定功能有关(图 3)。

图3 陆地棉MIKCC基因的基因结构和保守基序分析Fig.3 Phylogenetic relationship and gene structure analysis ofMIKCCgenes inG.hirsutum
2.3 陆地棉MIKCC家族基因的表达分析
为了研究MIKCC基因的表达特征,利用来源于陆地棉的RNA-seq数据分析了MIKCC基因在根、茎、叶、花托、萼片、雌蕊、雄蕊、花瓣、开花前 3 d、1 d、开花当天(0 d)、开花后 1 d、3 d、5 d、10 d、20 d、25 d 和 35 d 的胚珠以及开花后 5 d、10 d、20 d和25 d的纤维共22个组织器官及发育时期的表达量。通过表达聚类分析,发现MIKCC家族基因的时空表达分化明显。根据组织器官和发育时期表达的特征,MIKCC家族基因可分为3组。第1组基因在生殖器官和营养器官中表达,包含A,B和BS亚家族基因,主要为一些调控基因,调控萼片、花瓣及影响雄蕊形成。第2组基因在所有组织中都低水平表达,包含AG15,SOC1-like,SVP1-like,TM8亚家族, 这类基因主要参与控制开花时间。第3组基因不仅在花器官中表达,还在胚珠和纤维中持续表达,包含C/D和E亚家族,主要参与功能基因调控,如调控心皮、胚珠的发育及影响雄蕊形成。此类基因可能与棉花纤维的发育和生长有关。这些结果表明MIKCC基因在陆地棉中的功能分化(图4),为进一步针对性地研究不同基因的功能奠定了基础。

图4 MIKCC基因在陆地棉不同组织器官的聚类表达分析Fig.4 Expression cluster ofMIKCCgenes in different tissues and organs and different stages ofG.hirsutum
2.4 陆地棉MIKCC基因受miRNA调控的靶位点分析
作为转录因子,MIKCC调控靶基因的时空表达,同时其本身也受到严格的调控。前人研究发现,许多MADS-box基因是miRNA的靶标基因。比如,miR-444以AGL17类的mRNA为靶标[36],拟南芥中miR824能特异性降低AGL16的转录水平。本研究发现100个陆地棉MIKCC基因中12个基因具有miRNA靶基因位点(表1)。其中ANR1-like亚 家 族 基 因GhMADS19A,Gh-MADS19D,GhMADS51A和GhMADS51D含有miR7716的靶位点。控制植物开花转变的SOC1-like亚家族基因GhMADS21D和SVP-like亚家族基因分别为miR8677和miR1062的靶基因。此外,控制花器官发育的A亚家族基因Gh-MADS41A和E亚家族基因GhMADS14A/D也具有miRNA的靶位点,说明陆地棉MIKCC基因受到miRNA调控。
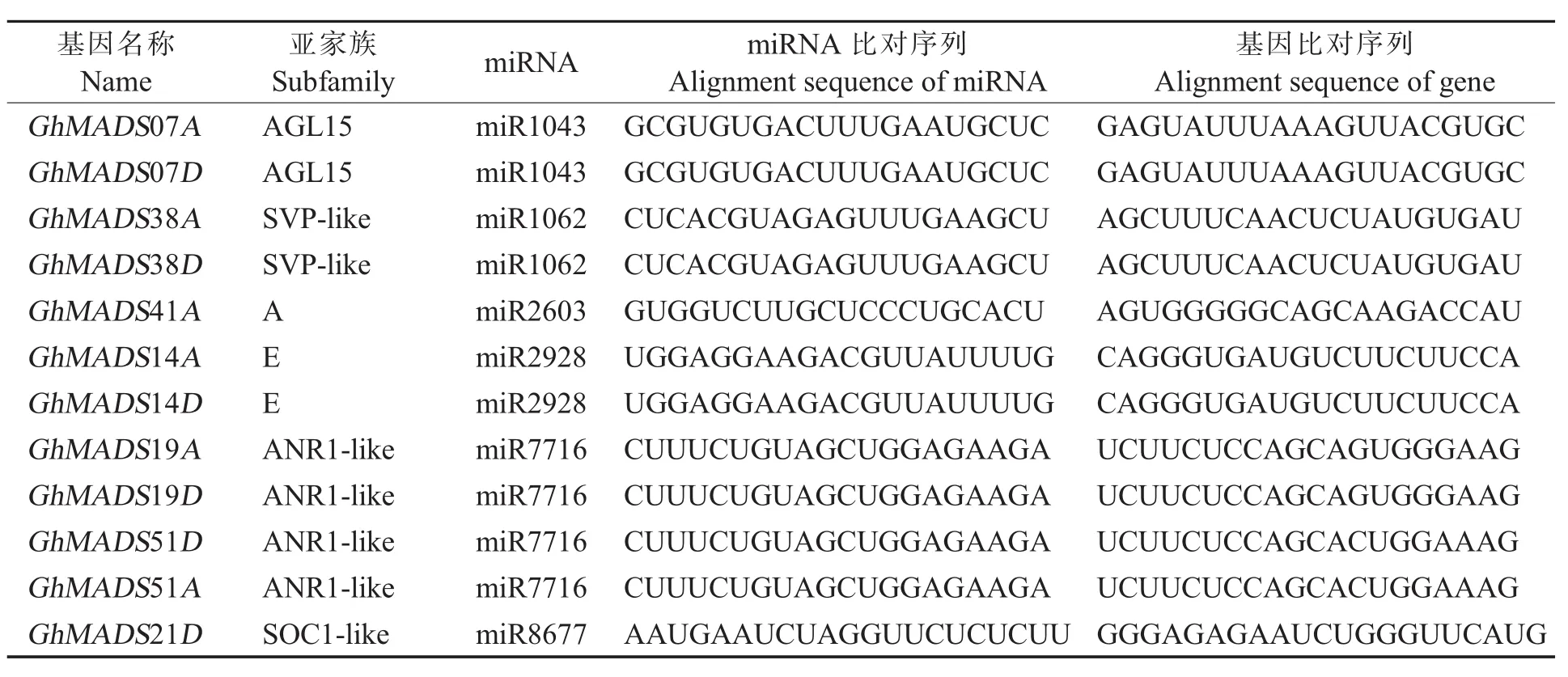
表1 陆地棉MIKCC基因受miRNA调控的靶位点分析Table 1 Analysis of the target sites ofMIKCCregulated by the corresponding miRNA inG.hirsutum
3 讨论
在四倍体棉花陆地棉TM-1的基因组中,我们鉴定了49对共线性的MIKCC型MADS-box基因,与杨树中MIKCC型MIKCC基因数量相近,多于拟南芥和葡萄[37]。这些基因被分为11个亚类,包含拟南芥中未发现的TM8类[3,38]。在拟南芥中,FLC亚族的基因控制春化和开花转变[39],但是,我们发现陆地棉TM-1基因组缺少FLC-like亚类基因,这与前人报道的结果一致[40],Jiang等[40]认为这可能与棉花种子不需要春化有关。在拟南芥中,除FLC-like控制开花转变的基因,还有SVP-like和SOC1-like亚家族[17,41-42]。我们发现,棉花中除了缺失FLC-like外,SOC1-like基因的数量也有所减少,但是SVP-like基因的数量出现扩增的现象,说明SVP-like亚家族基因可能在棉花开花转变控制中发挥重要作用。
棉花的花器官发育符合ABCDE开花模型,与拟南芥极为相似。与葡萄中A亚组的基因沉默不同[43],棉花中参与花器官发育的ABCDE亚家族基因均在花器官中特定的部位表达。很多证据表明MIKCC基因在棉花纤维发育中起重要作用[22,44]。Li等发现GhMADS11基因在棉花纤维细胞中积累,该基因可能参与棉纤维伸长[22]。Gh-MADS14在棉花纤维发育时期高水平表达,该基因可能通过GA信号参与棉花的纤维发育[44]。MIKCC基因除了在棉花的花器官中表达之外,C/D和E亚家族基因也在棉纤维发育时期高表达,特别是C/D亚家族基因,该亚组的4对基因在纤维发育过程中均高水平表达,在纤维发育过程中持续表达,说明该类亚家族基因不仅调控棉纤维伸长,还调控棉纤维的次生壁加厚。
拟南芥miR824能特异性降低AGL16的表达水平,从而影响叶片气孔的发育[45]。水稻的miR444通过它的靶标OsMADS23,OsMADS27a和OsMADS57调控OsRDR1的表达,从而影响水稻的抗病性[46]。在拟南芥中,ANR1通过硝酸盐信号参与侧根的生长。在水稻中,有4个基因与拟南芥的ANR1基因同源,这些基因受单子叶植物特有的miR444调控,不仅参与侧根的生长,并且介导氮的积累和磷饥饿下的响应[47]。在棉花中,ANR1-like亚家族基因GhMADS19A/D和Gh-MADS51A/D是miR7716的靶标,说明在陆地棉中,miR7716可能通过介导GhMADS19A/D和GhMADS51A/D的转录参与侧根的硝酸盐信号响应。拟南芥中的MADS-box基因AGL15和AGL18以及SVP和AGL24在生殖阶段对于阻碍花器官的形成至关重要[48]。有意思的是,棉花中AGL15亚家族基因GhMADS07A/D和SVP-like亚家族基因GhMADS38A/D都有miRNA的靶位点,这些结果说明陆地棉的开花转变可能受miRNA的调控,从而控制陆地棉的开花时间。
4 结论
以栽培种陆地棉TM-1为研究对象,在陆地棉的基因组中,共鉴定出100个棉花的MIKCC基因,并对这些基因进行了染色体分布和串联重复分析。基因结构分析发现,尽管该类家族基因非常保守,但是基因内含子的长度差异很大。表达分析发现,尽管棉花的MIKCC基因与其他物种类似,在花器官中表达,但是有些MIKCC基因在棉花纤维发育时期表达,显示出该类基因在棉花中的功能分化。此外,miRNA靶位点预测发现,很多MIKCC基因可能受miRNA调控,从而调控棉花生长发育。通过对陆地棉MIKCC基因的全方位分析,为棉花MIKCC基因的功能验证奠定了基础。
[1]Alvarez-Buylla E R,Liljegren S J,Pelaz S,et al.MADS-box gene evolution beyond flowers:Expression in pollen,endosperm,guard cells,roots and trichomes[J].Plant Journal,2000,24(4):457-466.
[2]Liu Y,Cui S,Wu F,et al.Functional conservation of MIKC*-type MADS box genes inArabidopsisand rice pollen maturation[J].Plant Cell,2013,25(4):1288-1303
[3]Kofuji R,Sumikawa N,Yamasaki M,et al.Evolution and divergence of the MADS-box gene family based on genome-wide expression analyses[J].Molecular Biology&Evolution,2003,20(12):1963-1977.
[4]Kaufmann K,Melzer R,Theissen G.MIKC-type MADS-domain proteins:Structural modularity,protein interactions and network evolution in land plants[J].Gene,2005,347(2):183-198.
[5]Henschel K A.Strukturelle und funktionelle charakterisierung von MADS-Box-genen aus dem laubmoosPhyscomitrella patens(Hedw.)B.S.G.[D].Cologne:University of Cologne,2002.
[6]Gramzow L,Thei覻en G.Phylogenomics of MADS-Box genes in plants-Two opposing life styles in one gene family[J].Biology,2013,2(3):1150-1164.
[7]Duan W,Song X,Liu T,et al.Genome-wide analysis of the MADS-box gene family inBrassica rapa(Chinese cabbage)[J].Molecular Genetics and Genomics,2015,290(1):239-255.
[8]Moon J,Suh S S,Lee H,et al.The SOC1 MADS-box gene integrates vernalization and gibberellin signals for flowering inArabidopsis[J].Plant Journal,2003,35(5):613-623.
[9]Tapia-López R,García-Ponce B,Dubrovsky J G,et al.An AGAMOUS-related MADS-box gene,XAL1(AGL12),regulates root meristem cell proliferation and flowering transition inArabidopsis[J].Plant Physiology,2008,146(3):1182-1192.
[10]Nesi N,Debeaujon I,Jond C,et al.The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentationoftheseedcoat[J].PlantCell,2002,14(10):2463-2479.
[11]Zahn L M,Leebens-Mack J H,Arrington J M,et al.Conservation and divergencein theAGAMOUS,subfamilyof MADS-box genes:evidence of independent sub-and neofunctionalization events[J].Evolution&Development,2006,8(1):30-45.
[12]Pa enicová L,Folter S D,Kieffer M,et al.Molecular and phylogenetic analyses of the complete MADS-Box transcription factor family inArabidopsis:New openings to the MADS world[J].Plant Cell,2003,15(7):1538-1551.
[13]Liu C,Chen H,Hong L E,et al.Direct interaction of AGL24 and SOC1 integrates flowering signals inArabidopsis[J].Development(Cambridge,England),2008,135(8):1481-1491.
[14]Ratcliffe O J,Kumimoto R W,Wong B J,et al.Analysis of theArabidopsisMADS AFFECTING FLOWERING gene family:MAF2 prevents vernalization by short periods of cold[J].Plant Cell,2003,15(5):1159-1169.
[15]Adamczyk B J,Lehtishiu M D,Fernandez D E.The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition inArabidopsis[J].Plant Journal,2007,50(6):1007-1019.
[16]Michaels S D,Amasino R M.FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering[J].Plant Cell,1999,11(5):949-956.
[17]Lee J H,Yoo S J,Park S H,et al.Role of SVP in the control of flowering time by ambient temperature inArabidopsis[J].Genes&Development,2007,21(4):397-402.
[18]Hu J Y,Meaux J D.miR824-regulated AGAMOUS-LIKE16 contributes to flowering time repression inArabidopsis[J].The Plant cell,2014,26(5):2024-2037.
[19]Yu F,Huaxia Y,Lu W,et al.GhWRKY15,a member of the WRKY transcription factor family identified from cotton(Gossypium hirsutumL.),is involved in disease resistance and plant development[J].BMC Plant Biology,2012,12(1):144.
[20]Zheng S Y,Guo Y L,Xiao Y H,et al.Cloning of a MADS box protein gene(GhMADS1)from cotton(Gossypium hirsutumL.)[J].Acta Genetica Sinica,2004,31(10):1136-1141.
[21]Guo Y,Zhu Q,Zheng S,et al.Cloning of a MADS box gene(GhMADS3)from cotton and analysis of its homeotic role in transgenicTobacco[J].Journal of Genetics&Genomics,2007,34(6):527-535.
[22]Li Y,Ning H,Zhang Z,et al.A cotton gene encoding novel MADS-box protein is preferentially expressed in fibers and functions in cell elongation[J].Acta Biochimica Et Biophysica Sinica,2011,43(8):607-617.
[23]Yang Z,Li C,Ye W,et al.GhAGL15s,preferentially expressed during somatic embryogenesis,promote embryogenic callus formation in cotton(Gossypium hirsutumL.)[J].Molecular Genetics and Genomics,2014,289(5):873-883.
[24]Zhang W,Fan S,Pang C,et al.Molecular cloning and function analysis of twoSQUAMOSA-Like MADS-box genes fromGossypium hirsutumL.[J].Journal of Integrative Plant Biology,2013,55(7):597-607.
[25]Zhang T,Hu Y,Jiang W,et al.Sequencing of allotetraploid cotton(Gossypium hirsutumL.acc.TM-1)provides a resource for fiber improvement[J].Nature Biotechnology,2015,33(5):531-537.
[26]Li F,Fan G,Wang K,et al.Genome sequence of the cultivated cottonGossypium arboreum[J].Nature Genetics,2014,46(6):567-72.
[27]Wang K,Wang Z,Li F,et al.The draft genome of a diploid cottonGossypium raimondii[J].Nature Genetics,2012,44(10):1098-1103.
[28]Finn R D,Clements J,Arndt W,et al.HMMER web server:2015 update.[J].Nucleic Acids Research,2015,43(1):30-38.
[29]Edgar R C.MUSCLE:Multiple sequence alignment with high accuracy and high throughput[J].Nucleic Acids Research,2004,32(5):1792-1797.
[30]Tamura K,Peterson D,Peterson N,et al.MEGA5:Molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance,and maximum parsimony methods[J].Molecular Biology&Evolution,2011,28(10):2731-2739.
[31]Altschul S F,Madden T L,Sch覿ffer A A,et al.Gapped BLAST and PSI-BLAST:A new generation of protein database search programs[J].Nucleic Acids Research,1997,25(17):3389-3402.
[32]Cox M P,Peterson D A,Biggs P J.SolexaQA:At-a-glance quality assessment of Illumina second-generation sequencing data[J].BMC Bioinformatics,2010,11(1):485.
[33]Trapnell C,Roberts A,Goff L,et al.Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks[J].Nature Protocols,2014,7(3):562-578.
[34]Dai X,Zhao P X.psRNATarget:a plant small RNA target analysis server[J].Nucleic Acids Research,2011,39(web server issue):155-159.
[35]Wang Y,Tang H,Debarry J D,et al.MCScanX:A toolkit for detection and evolutionary analysis of gene synteny and collinearity[J].Nucleic Acids Research,2012,40(7):e49.
[36]Gramzow L,Theissen G.A hitchhiker's guide to the MADS world of plants[J].Genome Biology,2010,11(6):214.
[37]Wei B,Zhang R Z,Guo J J,et al.Genome-wide analysis of the MADS-box gene family inBrachypodium distachyon[J].PLoS One,2014,55:245-256.
[38]Pnueli L,Abu-Abeid M,Zamir D,et al.The MADS box gene family in tomato:Temporal expression during floral development,conserved secondary structures and homology with homeotic genes fromAntirrhinumandArabidopsis[J].Plant Journal,1991,1(2):255-266.
[39]Müller F,Frentzen M.Phosphatidylglycerophosphate synthases fromArabidopsis thaliana[J].Febs Letters,2001,509(2):298-302.
[40]Jiang W,Chang S,Li H,et al.Liquid hot water pretreatment on different parts of cotton stalk to facilitate ethanol production[J].Bioresource Technology,2015,176:175-180.
[41]Gregis V,Sessa A,Colombo L,et al.AGL24,SHORT VEGETATIVE PHASE,andAPETALA1 redundantly control agamous during early stages of flower development inArabidopsis[J].Plant Cell,2006,18(6):1373-1382.
[42]Liu C,Zhou J,Brachadrori K,et al.Specification ofArabidopsisfloral meristem identity by repression of flowering time genes[J].Development,2007,134(10):1901-1910.
[43]Cubas P,Martínez-Zapater J M,Carmona M J.Floral meristem identity genes are expressed during tendril development in grapevine[J].Plant Physiology,2004,135(3):1491-1501.
[44]Zhou Y,Li B Y,Li M,et al.A MADS-box gene is specifically expressed in fibers of cotton (Gossypium hirsutum)and influences plant growth of transgenicArabidopsisin a GA-dependent manner[J].Plant Physiology&Biochemistry,2014,75(2):70-79.
[45]Kutter C,Sch觟b H,Stadler M,et al.MicroRNA-mediated regulation of stomatal development inArabidopsis[J].Journal of the American Statistical Association,2007,19(8):2417-2429.
[46]Wang H,Jiao X,Kong X,et al.A signaling cascade from miR444 to RDR1 in rice antiviral RNA silencing pathway[J].Plant Physiology,2016,170(4):2365-2377.
[47]Yan Y,Wang H,Hamera S,et al.miR444a has multiple functions inthericenitrate-signalingpathway[J].Plant Journal,2014,78(1):44-55.
[48]Fernandez D E,Wang C T,Zheng Y,et al.The MADS-domain factors agamous-like15 and agamous-like18,along with short vegetative phase and agamous-like24,are necessary to block floral gene expression duringthe vegetative phase[J].Plant Physiology,2014,165(4):1591-1603. ●
Genome-Wide Analysis ofMIKCCGene Family in Cotton
Zhou Na,Wang Luyao,Zhang Tianzhen,Hu Yan*
(State Key Laboratory of Crop Genetics and Germplasm Enhancement,Nanjing Agricultural University,Nanjing210095,China)
[Objective]In plants,floral organ identity and the process of the transition from vegetative to reproductive development is regulated byMIKCCgene family.Whole genome analysis is helpful to understand the evolutionary history ofMIKCCgenes and provides the basis for an in-depth analysis of molecular mechanism ofMIKCCfamily in flowering and floral organ development in cotton.[Method]Whole genome identification ofMIKCCgenes were carried by HMMER 3.0 and pfam gene bias expression and were clustered based on the RNA-seq data.[Result]We identified 100MIKCCgenes and classified them into 11 subfamilies,according to their phylogenetic relationships to theArabidopsisandVitis vinifera MIKCCgenes.According to their expression profiles,MIKCCgenes were classified into four types.TheMIKCCgenes of C/D and E subfamily were particularly highly expressed during cotton fiber elongation stage,indicating gene functional differentiation in cotton evolution.12 of the 100MIKCCgenes contains miRNA target sites,indicating that the flower development and the transition from vegetative to reproductive development of cotton may be regulated by miRNAs.[Conclusion]This study not only showed the functional differentiation ofMIKCCgenes in cotton fiber development,but also found thatMIKCCfamily genes may be regulated by miRNAs in the growth and development of cotton,which provided the useful information for further functional studies ofMIKCCgenes in cotton.
upland cotton;MIKCCgene;functional differentiation;miRNA
S562.03 文献标志码:A
1002-7807(2017)06-0495-09 DOI:10.11963/1002-7807.znhy.20170913
2017-04-17
周娜(1990―),女,硕士,2014101153@njau.edu.cn。 *通信作者:njauhuyan@163.com
中央高校基本科研业务费自主创新重点研究项目(2015001)
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
流行色(2021年8期)2021-11-09
实用肿瘤学杂志(2020年4期)2020-12-08
汉语世界(The World of Chinese)(2019年5期)2019-11-11
少儿科学周刊·少年版(2018年12期)2018-01-26
科学中国人(2017年36期)2017-06-09
上海农业学报(2017年3期)2017-04-10
红领巾·探索(2015年9期)2015-09-10
中国老年学杂志(2015年9期)2015-01-31
植物营养与肥料学报(2014年1期)2014-03-11
