
基因组编辑脱靶研究进展
2017-11-21 00:48何秀斌谷峰
生物工程学报 2017年10期
何秀斌,谷峰
基因组编辑脱靶研究进展
何秀斌,谷峰
温州医科大学眼视光学院眼视光学和视觉科学国家重点实验室,浙江温州 325000

何秀斌, 谷峰. 基因组编辑脱靶研究进展. 生物工程学报, 2017, 33(10): 1757–1775.He XB, Gu F. Genome-editing: focus on the off-target effects. Chin J Biotech, 2017, 33(10): 1757–1775.
近年各种基因组编辑技术的成功研发为人类疾病的治疗与预防谱写了新的篇章,这些技术对应的基因组编辑工具主要包括锌指核酸酶 (ZFNs)、转录激活子样效应因子核酸酶 (TALENs) 和最近发现的规律成簇间隔短回文重复(CRISPR)/Cas系统。这些工具相应的脱靶问题目前是制约基因组编辑技术介导人类疾病治疗的重要瓶颈。本文将分别从基因组编辑工具的介绍、脱靶的现状、解决优化的方案和检测方法进行总结与探讨,通过比较,进一步了解基因组编辑工具的优缺点及相关脱靶检测方法的适用性。
基因组编辑,脱靶,锌指核酸酶 (ZFNs),转录激活子样效应因子核酸酶 (TALENs),规律成簇间隔短回文重复 (CRISPRs)
随着人类基因组计划的实施,人类对严重危害健康的疾病有了更加全面和深入的认识。基因治疗 (Gene therapy) 这一概念自问世以来,正在逐渐改变人类疾病的治疗方式。基因治疗尤其为单基因遗传病的患者带来了福音[1]。传统的基因治疗是利用野生型基因去补偿突变基因的功能,但是仍然存在转基因沉默和随机插入等问题。基因组编辑工具的成功研发为基因的原位修复提供了可能。基因组编辑工具包括锌指核酸酶 (Zinc-finger nucleases,ZFNs)、转录激活子样效应因子核酸酶 (Transcription activator-like effector nucleases,TALENs) 和最新发现的规律成簇间隔短回文重复和Cas蛋白的DNA核酸内切酶系统(Clustered regulatory interspaced short palindromic repeat (CRISPR)/Cas- based RNA-guided DNA endonucleases)。然而,这3种工具酶在识别并切割靶位点的同时,也对与靶位点相似的DNA序列同样进行切割,即脱靶 (Off-targets)。脱靶可能破坏细胞内的重要基因,从而导致其他疾病,脱靶问题是基因组编辑工具在临床应用受限的重要瓶颈之一[2]。本文将对这3种基因组编辑工具的脱靶现状、解决脱靶的方案、相关的脱靶检测方法等问题进行总结,以期更好地解决该重大科学问题。
1 基因组编辑工具的脱靶现状及解决方案
1.1 锌指核酸酶 (ZFNs)
锌指核酸酶 (ZFNs) 为第一代应用于基因组编辑的核酸酶,它由结合DNA的锌指蛋白结构域和非特异性的核酸内切酶Ⅰ结构域融合而成,其中Ⅰ源于海床黄杆菌,是一种人工设计的DNA限制性内切酶[3]。目前已成功在拟南芥、烟草、秀丽隐杆线虫、黑腹果蝇、斑马鱼、非洲爪蟾、大鼠、小鼠和人类细胞 (包括体细胞、胚胎干细胞、诱导性多能干细胞) 等中进行基因组编辑[4]。通过在靶序列两侧设计特定的锌指蛋白识别和结合DNA,引导Ⅰ二聚体在两结合位点之间进行剪切产生双链断裂 (Double-strand breaks,DSBs) 来实现同源重组修复 (Homology-directed recombination,HDR) 或非同源末端连接 (Non-homologous end joining,NHEJ),从而实现基因组特定位点的编辑[6]。锌指核酸酶编辑的靶向性与锌指蛋白的DNA识别特异性、靶位点序列和锌指核酸酶的转运方式等有关,但大部分取决于负责识别和结合DNA的锌指蛋白[4,6]。一般每个锌指模块识别3−4个碱基 (Base pair,bp),而其识别DNA长度的限制性降低了锌指核酸酶靶向编辑的特异性 (图1),通常需要通过设计多个模块来提高识别的特异性[7],但仍然存在脱靶问题,在Marina Bibikova等对果蝇的基因编辑时首次被发现[5],随后又在编辑斑马鱼和基因、人和基因中被检测到[8-10]。
针对影响锌指核酸酶脱靶的相关因素,研究者们采取了多种策略和方法来提高其特异性。在靶序列设计方面,可以运用在线的生物信息设计工具PROGNOS (Predicted report of genome-wide nuclease off-target sites),通过预测的脱靶情况来选择最佳的靶序列以最大限度减少脱靶[11-12](表1)。在锌指蛋白设计方面,锌指模块设计的个数越多,识别的序列就越长,相应序列在基因组中的唯一性也越强;另外有研究者通过设计异源的锌指蛋白二聚体来降低脱靶的发生率,相较于同源锌指蛋白二聚体,异源二聚体的相互作用会减弱,而与靶DNA位点的结合能力就相对增强,且只有当形成异源二聚体时才能发挥作用,减少了脱靶的可能性[13-14]。对于负责切割的非特异性Ⅰ内切酶,也有研究者对其进行改造,使其只切割一条链,在增加HDR效率的同时降低了脱靶效应[15]。在锌指核酸酶的转运方式方面,Gaj等直接将锌指核酸酶以蛋白的形式转入细胞,显示高效的编辑效率和较小的脱靶率[16]。

图1 ZFNs的脱靶示意图
1.2 转录激活子样效应因子核酸酶 (TALENs)
转录激活子样效应因子核酸酶 (TALENs) 的结构域组成和作用模式与锌指核酸酶相似,通过转录激活子样效应因子 (TALEs) 识别和结合DNA,引导Ⅰ在靶位点产生DSB,同样通过HDR或NHEJ方式完成基因组编辑。TALEs最早在植物致病菌黄单胞杆菌中发现,包含易位结构域、DNA结合结构域、核定位信号 (Nuclear localization signals,NLS) 和转录激活结构域 (Transcriptional activate domain),其中DNA结合结构域是决定TALENs特异性的重要部分,它由1.5−33.5个串联重复序列 (Tandem repeats,TRs) 组成,每个重复包含33−35个氨基酸,而12和13位的氨基酸是可变的,能够识别特定的碱基序列,因此这两个氨基酸被称为重复变异双残基 (Repeat variablediresidues,RVDs)[17]。TALENs目前已成功编辑了酵母、果蝇、水稻、人类体细胞和胚胎干细胞等微生物、植物和动物细胞中的内源性基因[18],并且成功用于制造疾病细胞模型和动物模型[19-20]。相较于ZFNs,TALENs编辑效率与之相当,但是脱靶率较低[21],很大部分原因在于其每个串联重复序列只识别一个碱基[22-23](图2),而ZFNs的一个锌指模块识别3−4个碱基,在精确度上,TALENs更胜一筹。
尽管脱靶率较低,但是若应用于临床疾病的基因治疗,理论上需做到无脱靶,以期减少对人体的毒性作用。因此为了进一步提高TALENs编辑基因的特异性,研究者们也提出了相应的解决策略。第一,运用生物信息学专业在线工具设计和选择脱靶率低的靶序列,如CHOPCHOP、PROGNOS和TALE-NT 2.0[11,24-26](表1)。第二,TALENs的重复长度会影响特异性,偏短的TALENs对每个碱基识别的特异性更高,偏长的TALENs对整个靶序列识别的特异性更高,这与结合DNA所需要的能量有关,较短的TALENs结合DNA所需的能量较少,对应识别每个碱基所分布到的能量就多,特异性也就越强,反之则越低;此外TALENs的作用浓度过高使靶位点饱和,使之倾向于结合脱靶位点,也会降低特异性,因此设计合适的长度、采用合适的浓度是降低脱靶率的关键[27]。第三,通过获得TALEs的变体筛选高特异性的TALE,结果显示,改变C端的结构域 (K788Q、R792Q和R801Q) 可减少阳离子电荷量,能提高特异性至野生型的10倍[27];缩短C端残基的数目,可显著地降低脱靶率[28-30];改变N端的结构域,使之倾向特异识别胸腺嘧啶 (Thymine)[31-32]。第四,将TALE与其他特异性核酸内切酶如Ⅰ-Ⅰ和Ⅰ-Ⅰ嵌合,可提高识别的特 异性[33]。
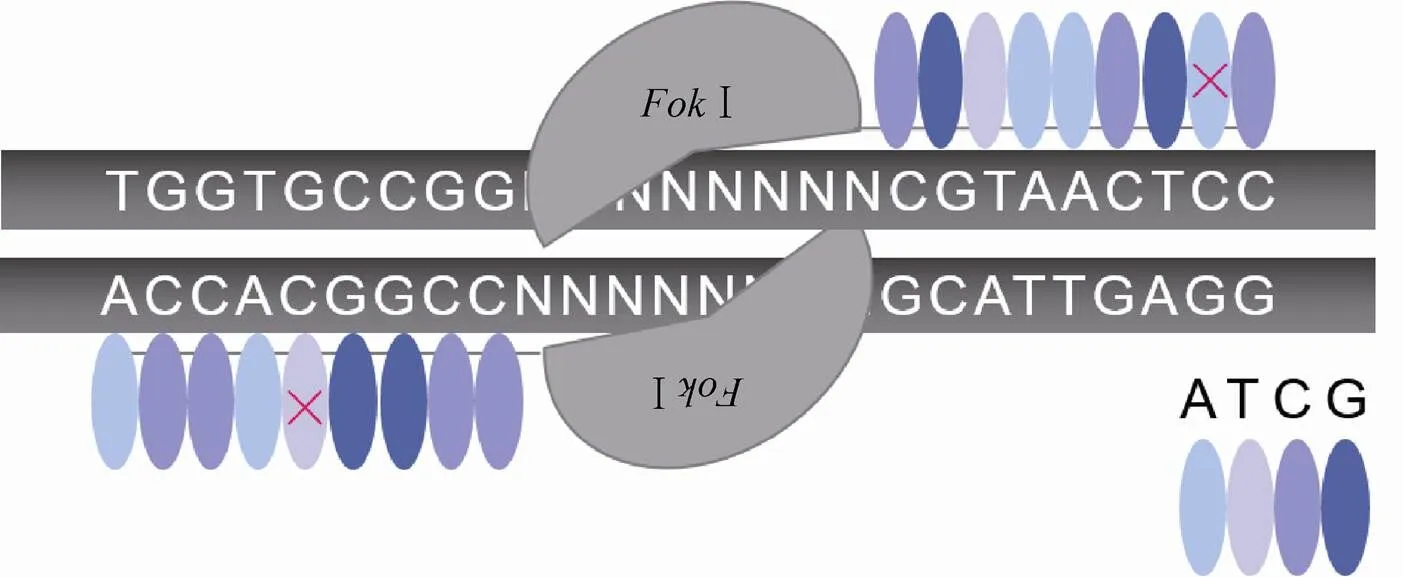
图2 TALENs的脱靶示意图
1.3 规律成簇间隔短回文重复(CRISPR)/Cas核酸酶
CRISPR/Cas系统为细菌和古细菌中的免疫防御系统,目前最常用于基因组编辑的为CRISPR/Cas9和CRISPR/Cpf1系统,分别属于CRISPR 2类系统中的Ⅱ型和Ⅴ型[[34]。CRISPR/ Cas9系统(图3) 由Cas9蛋白 (包含HNH和RuvC两个结构域)、crRNA和tracrRNA组成,经改造,crRNA和tracrRNA可设计为单个向导RNA (Single guide RNA,sgRNA) 引导Cas9蛋白靶向切割DNA产生DSB,并由HDR或NHEJ方式介导修复,识别靶位点主要由互补的sgRNA和3′端的PAM (Protospacer adjacent motif) 序列决定,不同来源的CRISPR系统的PAM序列不尽相同[35]。最先用于哺乳动物细胞编辑的是来源于化脓性链球菌的CRISPR/SpCas9系统,研究者采用20 nt (Nucleotide) 的sgRNA靶向带有NGG的PAM序列,成功对内源性基因进行了编辑[36]。而后发现的来源于金黄色葡萄球菌的CRISPR/SaCas9、脑膜炎奈瑟菌的CRISPR/NmCas9、嗜热链球菌的CRISPR/StCas9和最新发现来源于空肠弯曲菌的CRISPR/CjCas9都可成功用于哺乳动物细胞的基因组编辑,但是均存在脱靶现象,甚至发现部分脱靶位点的编辑效率与靶位点相当或高于靶位点[37-43]。

图3 CRISPR/Cas9的脱靶示意图
通过研究发现有因素会影响CRISPR/Cas9系统的特异性,包括靶位点的选择、Cas9蛋白、sgRNA的长度、转运方式及小分子化合物等。选择脱靶率低的靶位点是基因组编辑的第一步,可采用一系列在线工具对靶位点进行脱靶评估和筛选[44](表1)。研究发现SpCas9对其他PAM序列 (NGA和NAG) 也有编辑的活性,间接说明了CRISPR/Cas9脱靶问题,因此另有研究者通过结构分析、组合设计和细菌选择系统筛选了针对NGA、NGAG和NGCG PAM序列的突变SpCas9,显示在部分位点具有更高的效率和特异性[45-46]。在sgRNA的设计方面,就SpCas9而言,17 nt或18 nt的截短sgRNA能减少脱靶,由于sgRNA与DNA结合所需的能量减少,相对地提高了特异性,这与TALENs的长度设计原理相似,且与Cas9n组合可进一步提高特异性[47]。在转运方式上,将Cas9蛋白和sgRNA与核糖核蛋白 (Ribonucleoproteins,RNPs) 融合成复合物直接导入细胞,而非将质粒导入细胞,结果显示其能提高基因组编辑的特异性,由于是将Cas9蛋白导入细胞,其作用时间受到了一定的限制,不会像质粒持续表达Cas9蛋白,进而减少其他脱靶位点的作用[48-52],该方法已经在治疗年龄相关性黄斑变性疾病模型中取得了一定成效[53]。对于Cas9蛋白,研究者们对其进行了各种不同的改造,麻省理工大学的张锋组研究显示,采用双切口的Cas9 nickases (Cas9n) 对哺乳动物细胞和小鼠受精卵进行编辑,与野生型Cas9相比具有更高的特异性[35],而利用单个的Cas9n对牛受精卵进行基因敲入发现,脱靶率与野生型Cas9相比较低,并且受精卵存活率也大幅度升高[54]。许多研究者不限于Cas9本身的突变,对其结构也进行了许多改造来提高特异性,比如内含肽灭活的Cas9系统,Cas9突变体带有雌激素受体结合域,只有当4-羟基他莫昔芬 (4-hydroxytamoxifen,4-HT) 与雌激素受体结合后,Cas9才能被激活进行基因组编辑[55];类似的系统还有光激活的Cas9系统[[56-58]、分离的Cas9突变体系统[59-60]、小分子诱导的Cas9系统[61]和变构调节的Cas9系统[62],都能不同程度地降低脱靶率。最直接的是获得高保真的Cas9突变体eSpCas9和SpCas9-HF1,通过降低Cas9蛋白与DNA糖磷酸骨架的非特异性相互作用,从而降低脱靶率[63-64]。此外,有研究者将失活的Cas9 (Dead Cas9,dCas9) 与Ⅰ融合,形成dCas9-Ⅰ系统以二聚体形式作用靶位点,也能提高特异性,dCas9虽然失去了活性,但是还保留与DNA结合的能力[65-67]。以上降低脱靶的策略理论上可以相互联合,协同作用增加CRISPR/Cas9系统的靶向特异性。
CRISPR系统以往一直针对基因片段进行编辑,而目前有研究者们将CRISPR/Cas9与激活诱导的胞苷脱氨酶 (Activation induced-cytidine deaminase,AID) 或相应的同源基因 (APOBEC1和 PmCDA1等) 联合用于编辑单个碱基,希望应用于点突变的原位修复。有研究者将之与nCas9或dCas9联合,用于靶向单碱基的编辑 (Cytosine→Thymine,C→T),发现脱靶率相较于Cas9低,Komor等发现dCas9-APOBEC1的编辑在预测脱靶位点未发现脱靶现象[68];Nishida等在nCas9-AID和dCas9-AID介导的单碱基编辑中检测到小于1.5%的脱靶率[69];Hess等发现利用两个MS2发夹结构修饰的sgRNA与dCas9-AID作用能减少脱靶[70];Kim等为降低nCas9-APOBEC1的脱靶率,采用改造的Cas9 (针对不同PAM的Cas9突变体),结果显示脱靶率平均下降了3.6倍[71];Zong等用nCas9-APOBEC1和dCas9-APOBEC1编辑水稻在预测位点未发现脱靶[72];Kim等将nCas9-rAPOBEC1用于编辑小鼠胚胎,成功制造无义突变的疾病模型,且未在该模型中检测到其他突变位点,即脱靶位点[73]。目前从以上研究结果来看,针对单碱基编辑的脱靶率要比单用Cas9低很多,这对于点突变的基因纠正、疾病模型的建立和育种具有推动性的作用,但仍需要更多的实验来进一步验证。
CRISPR/Cpf1系统是近年最新发现的基因组编辑系统,只需要一个类似RuvC的Cpf1结构域和单个crRNA即可进行基因组编辑,目前发现的有来源于氨基酸球菌属 (sp. Cpf1,AsCpf1)、毛螺科菌 (Cpf1,LbCpf1) 和弗朗西斯菌属 (Cpf1,FnCpf1) 等多种Cpf1,与Cas9不同,Cpf1的PAM序列在5′端,其小型和便捷的特点在基因组编辑应用上更具有优势[74]。CRISPR/Cpf1最先在小鼠上进行基因组编辑,Kim等采用AsCpf1和LbCpf1对小鼠受精卵进行基因敲除,靶向深度测序 (Targeted deep sequencing) 结果显示在2−4 bp的sgRNA错配序列中未发现脱靶现象,但在1 bp错配时存在约1/6的脱靶率[75],同时Hur等将Cpf1与RNP组装同样编辑小鼠受精卵,采用全基因组测序 (Whole genome sequencing,WGS) 检测发现在7 bp及以上的错配中未发现脱靶现象[76]。随后,研究者们在植物上进行了基因组编辑,在编辑大豆和烟草时,通过靶向深度测序在4 bp及以上的错配中未检测到脱靶[77],另有研究者对水稻进行编辑时在预测脱靶位点未检测到脱靶[78-79]。有研究运用多种脱靶检测方法 (Digenome-seq、GUIDE-seq和靶向深度测序) 比较了Cas9与Cpf1的脱靶率,结果显示Cpf1在人类细胞编辑的特异性高于Cas9[80-81],因此有望设计高效率的Cpf1突变体或复合物应用于基因治疗。同时,是否可以通过设计双切口酶的Cpf1,或进一步与Ⅰ融合形成高保真的Cpf1,来提高其编辑的特异性。
2 脱靶的检测方法及优缺点比较
2.1 预测脱靶位点的检测方法
用CasFinder等多款生物信息在线工具预测脱靶位点[82](表1),筛选出脱靶可能性高的位点进行检测,将包含预测脱靶位点的片段进行富集鉴定,如PCR扩增后使用Sanger测序、多重PCR扩增或液相探针特异性杂交富集脱靶片段后,进行二代测序 (Next generation sequence,NGS)。Sanger测序简便易行,不需要特殊的设备或技术,当样本量大时比较耗时且不经济,对于脱靶率低的位点也不能完全检测出。NGS成本虽然相对昂贵,但是在样本量多或者脱靶效率较小 (提高测序深度) 的情况下,通过多重PCR或液相探针杂交的方法将这些含候选脱靶位点的片段扩增出来,进行深度测序更值得考虑。也可将PCR产物进行T7E1或Surveyor检测 (图4),T7E1对插入缺失 (Indels) 比较敏感,而Surveyor对单核苷酸突变 (SNPs) 和小片段的indels相对敏感,因而T7E1检测DSBs产生的NHEJ更为敏感,但两者均容易受温度、时间、DNA与酶的比例和缓冲液盐离子浓度的影响[83],优点在于快速简便,但灵敏度低。由于是对预测的脱靶位点进行检测,可能忽略了其他的脱靶位点,因此存在一定的偏倚性,不能全面反映细胞内脱靶情况。
2.2 全外显子测序与全基因组测序
全外显子测序 (Whole exon sequencing,WES)和全基因组测序 (Whole genome sequencing,WGS),顾名思义,前者针对基因组的所有外显子,后者则没有选择性地对全基因组进行检测。两者比较,后者能直接真实地反映脱靶的情况,前者有可能忽略了非编码区的脱靶,而非编码区也是对基因起调控作用的重要区域。研究者们已在诱导性多能干细胞、秀丽隐杆线虫、疟原虫和拟南芥等多个物种中应用全基因组测序检测基因组编辑的脱靶情况[84-87]。无论是SNPs和indels,还是染色体水平的变化如易位、倒置等,WGS都能检测到,但对于大量样本中的低频脱靶位点仍然难以检测[88](由于测序成本等原因,WGS一般测序深度在30X−50X)。为优化脱靶的检测方案,以下其他的相关检测方法将通过富集脱靶位点,结合WES或WGS来研究脱靶。
2.3 ChIP-seq
染色体免疫共沉淀 (Cross-linking chromatin immunoprecipitation,ChIP) 技术可以针对性地获得蛋白质与DNA结合的复合物,与二代测序技术联合则可以全面地检测与蛋白质结合的DNA序列,该方法称为ChIP-seq[89]。在CRISPR/Cas9的脱靶检测中,利用失活的Cas9即dCas9与DNA序列进行结合而不发生切割的特点,从而确定Cas9的具体结合位点,包括靶位点和脱靶位点。将结合了dCas9的基因组碎片化,通过ChIP富集含有dCas9的DNA片段,再经过纯化获得DNA进行PCR和二代测序(图5)。研究者利用ChIP-seq对12个sgRNAs的脱靶进行检测,发现了10–1 000个不等的脱靶位点,基本位于染色质开放区域,且相当一部分与预测的脱靶位点不一致[39]。该技术也存在假阳性率,由于ChIP倾向结合高表达聚合酶Ⅱ、聚合酶Ⅲ和tRNA基因的区域[90],另外,与DNA结合和对DNA切割的位点可能存在差异。

图4 偏倚性预测和检测脱靶的方法
2.4 IDLV捕获
IDLV,即整合缺陷的慢病毒载体 (Integrative- deficient lentiviral vectors,IDLV),它们以游离的DNA形式存在于细胞核中,能够整合到发生DSBs的位点,利用该特点首次应用于ZFNs的脱靶检测中[91]。不同于完整的慢病毒载体,IDLV对基因组的整合没有偏好性,以往利用其检测病毒载体的安全性。当核酸酶切割基因组产生DSBs时,IDLV会在NHEJ修复期间整合于靶位点和脱靶位点,相当于“标记”了DSBs位点。其次提取细胞基因组并破碎,并在片段两头加上接头序列,因IDLV两端含有两个已知的LTR (Long terminal repeats) 序列,可以利用线性扩增PCR (Linear amplification-mediated PCR,LAM-PCR) 技术扩增位于IDLV两侧的片段,该技术常用于扩增未知的DNA序列,最后进行二代测序(图6)。随后该技术也应用于TALENs和CRISPR/Cas9,可以检测到1%的脱靶率,识别1–13 bp的错配[92-93]。使用该技术的优势在于IDLV能够高效地进入细胞核,包括难以转染的人类细胞,但同时也会整合到其他非核酸酶引发的DSBs位点,增加了假阳性率,因此需要做好对照。另外,该方法是否能够捕获到所有的脱靶的位点并不清晰。
2.5 BLESS
BLESS (Directbreaks labeling,enrichment on streptavidin,and next-generation sequencing) 技术是基于生物素-链亲和素原理设计的[94]。首先提取完整的细胞基因组,在体外利用核酸酶进行切割,后采用高特异性T4 DNA连接酶将生物素化的寡核苷酸与DSBs位点连接,该连接酶只能连接双链断裂的位置,形成发夹样结构。其次将基因组消化成片段,因生物素化和链亲合素化的寡核苷酸都含有相同的酶切位点,所以用相同的酶切后进行连接以达到富集的目的。经过纯化,并在发夹结构的另一端引入带酶切位点的寡核苷酸连接双链的断端,最后将片段的两头消化形成用于扩增的开放双链模板,再进行PCR和二代测序 (图7)。有研究者在对小鼠和人类细胞的基因组编辑时运用BLESS和ChIP-seq检测脱靶,结果显示BLESS能检测出更多的脱靶位点[41,64]。虽然该技术是对原位DSBs进行直接的检测,但由于是在体外进行的,可能不能真实地反映体内的情况。

图5 非偏倚性脱靶检测方法之ChIP-seq

图6 非偏倚性脱靶检测方法之IDLV捕获
2.6 GUIDE-seq
GUIDE-seq (Genome-wide,unbiased identification of DSBs enabled by sequencing) 技术与IDLV捕获技术原理相似,体内的细胞基因组在核酸酶作用下发生DSBs,此时引入“标签”——双链寡脱氧核苷酸 (Double-stranded oligodeoxynucleotides,dsODNs),该“标签”会整合到DSBs位点,然后提取细胞基因组随机打断成片段,在片段两头加上接头,因引入的dsODNs序列和接头序列已知,故对正反链分别进行LAM-PCR,再通过二代测序即可检测脱靶位点(图8),该方法同样能检测到比ChIP-seq更多的脱靶位点,敏感性小于0.1%[40]。与ChIP-seq、IDLV捕获和BLESS技术相比,GUIDE-seq相对简便,因此也有一些研究者以此方法检测脱靶,但只有在发生DSBs后立即引入dsODNs才能被检测[46,63,95]。另外,“dsODNs标签”是否都能够整合到细胞所有的DSBs并不清晰,IDLV捕获方法也有类似的问题。
2.7 LAM-HTGTS
LAM-HTGTS (Linear amplification-mediatedhigh-throughput genome-wide translocation sequencing)技术发展于检测AID引发的基因重排,建立在位点发生DSBs的基础上[96]。之后有研究者利用DSBs引发的重排原理来检测CRISPR/Cas9和TALEN进行基因组编辑的脱靶情况,细胞基因组在核酸酶作用下发生DSBs,引发基因重排,将提取的细胞基因组破碎,针对某个基因的脱靶,在该基因sgRNA附近设计带有生物素标签的捕获引物,通过LAM-PCR、生物素-链亲和素系统富集带有靶基因片段的序列,包括发生了重排和未发生重排的序列,再利用二代测序进行分析[97](图9)。该方法不需要引入额外的特殊序列,灵敏度比IDLV高,背景值比BLESS低,且相对经济,缺点在于只能检测发生重排的位点,而重排的概率比较低,约200–1 000个细胞中发生一次[98]。

图7 非偏倚性脱靶检测方法之BLESS

图8 非偏倚性脱靶检测方法之ChIP-seq
2.8 Digenome-seq
Digenome-seq (Cas9-digested whole genome sequencing) 是完全在体外进行的脱靶检测技术[99-100]。首先提取细胞基因组并消化片段,在片段两端加上相同的接头序列,而后将Cas9蛋白和sgRNA与消化的片段混合进行切割,这些片段中部分被切割,部分未被切割,再利用二代测序读取所有的片段并与基因组序列进行比对(图10)。该方法灵敏度高,能检测到0.1%的indels,且不限制于染色质结构影响,实验的可重复性和均一性也较高。Digenome-seq是先经过基因组的破碎,再进行核酸酶的切割,而前面有几个检测的方法都是先进行核酸酶的切割引入各种“标签”,再进行基因组的破碎,因此该技术是直接检测切割的位点而不是检测结合的位点,脱靶位点的覆盖范围相对较大,灵敏度也相对较高。采用“标签”引入的方式存在效率问题,这些“标签”并不是都能整合到DSBs的位点,尤其是对于低频脱靶位点。Digenome-seq的缺点在于进行二代测序分析时没有对发生切割和未发生切割的片段进行分离,故所需要的读长量大才能检测到低频的位点,最多需要400 million,并且它是完全在体外进行的脱靶检测技术,所以可能不能真实地反映体内的情况,由于不受染色质结构的影响,可能存在一定的假阳性率。
2.9 CIRCLE-seq
CIRCLE-seq (Circularization forreporting of cleavage effects by sequencing) 技术是研究人员最新开发的一种通过测序体外检测切割效应的方法[101]。同样是体外进行的实验,将基因组剪切成片段,在片段两端加上茎环结构的接头序列,通过酶切接头序列和分子内连接,使片段环化后用核酸酶切割,能被切割的环形片段都将线性化,不能被切割的维持环状结构,在线性化的片段上再次加上接头序列,然后进行PCR和NGS进行检测(图11)。该技术的优点在于它的读长量少 (4−5 million) 且随机读长背景低,而Digenome-seq的读长量最多需要400 million,并且CIRCLE-seq能检测到小于0.1%的脱靶位点。研究人员以靶向基因为例,显示CIRCLE-seq能鉴定出Digenome-seq已检测到的29个位点中的26个,并且还检测到156个其他方法无法检测的新位点。但由于该技术是在体外进行的,同样可能不能真实地反映在体内的情况。

图9 非偏倚性脱靶检测方法之LAM-HTGTS

图10 非偏倚性脱靶检测方法之Digenome-seq
2.10 SITE-seq
SITE-seq (Selective enrichment and identification of tagged genomic DNA ends by sequencing) 技术是与CIRCLE-seq同期出现的一项新的脱靶检测方法[102],也是体外进行的方法。首先获取高分子量的细胞基因组,用Cas9/RNA复合体 (sgRNPs) 体外切割基因组,在切割的位点加上生物素化的接头序列,然后进行基因组的破碎,再利用生物素进行富集,得到的是被核酸酶切割过的片段,在该片段的另一端加上接头序列后,进行PCR和二代测序分析 (图12)。与HTGTS和Guide-seq相比,SITE-seq不依赖于核酸酶的转运方式、细胞的类型和DNA的修复,但是SITE-seq检测到的位点比另外两者要多;相比于Digenome-seq,该技术能将脱靶位点进行富集并检测,提高了检测的灵敏度。在该技术中,检测到的脱靶位点的数量与sgRNPs的使用浓度相关,低浓度时检测到的脱靶位点少,相反高浓度时检测到的数量多,在一定浓度时能覆盖利用体内脱靶检测方法检测到的位点。利用该技术可以定位编辑活性高的基因组区域,指导选择活性和特异性最高的位点来减少脱靶效应。缺点在于SITE-seq是完全在体外进行,随着sgRNPs浓度的增加,其检测到的脱靶位点远多于体内检测到的位点,所以可能不能真实地反映细胞内的脱靶情况,但可以为编辑位点的选择提供指导依据。
3 展望
当前的ZFNs、TALENs和CRISPRs这三大类基因组编辑技术给基因治疗带来了极具潜能的应用前景,其中ZFNs的脱靶率比另两者都要高,即使经过改造以提高特异性。由于锌指结构的固有识别特点,使得其可改造的空间小于后两者。如果比较未经改造的TALENs和CRISPR系统,TALENs的特异性要高于CRISPR系统,但由于其设计和技术的复杂性限制了它的广泛使用,因此CRISPR系统的相对简便性和可改造性使其在基因组编辑领域处于优势和领先地位。尽管存在一系列脱靶问题,研究者们也针对该问题提出了多种解决的策略和方案,比如双切口的nCas9[36]、高保真的Cas9[63-64]、Cas9与小分子结合的系统[56-58,61-62]、dCas9-AID或nCas9-AID系统[68-69,71]等。利用相对合适的脱靶检测技术 (各脱靶检测技术的比较见表2) 和利用现有的方法最大限度地改善脱靶问题,降低其对细胞产生的毒性作用,以期将基因组编辑技术应用于临床的疾病治疗,从根本改善或治愈相关的遗传或非遗传性疾病。

图11 非偏倚性脱靶检测方法之CIRCLE-seq

图12 非偏倚性脱靶检测方法之SITE-seq


表2 各脱靶检测方法的比较
a, bThe quantity of $ or * represents the extent of corresponding strategy’s cost, time & complexity.cdetection means using the nucleases to cut the genome in living cells and then perform the off-target detection.detection means using the nucleases to cut the genomic DNAand then perform the off-target detection.
[1] King A. Gene therapy: a new chapter. Nature, 2016, 537(7621): S158–S159.
[2] Fruman DA, O’Brien S. Cancer: a targeted treatment with off-target risks. Nature, 2017, 542(7642): 424–425.
[3] Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA, 1996, 93(3): 1156–1160.
[4] Jo YI, Kim H, Ramakrishna S. Recent developments and clinical studies utilizing engineered zinc finger nuclease technology. Cell Mol Life Sci, 2015, 72(20): 3819–3830.
[5] Bibikova M, Golic M, Golic KG, et al. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics, 2002, 161(3): 1169–1175.
[6] Cornu TI, Thibodeau-Beganny S, Guhl E, et al. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther, 2008, 16(2): 352–358.
[7] Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol, 2016, 428(5): 963–989.
[8] Meng XD, Noyes MB, Zhu LJ, et al. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol, 2008, 26(6): 695–701.
[9] Gupta A, Meng XD, Zhu LJ, et al. Zinc finger protein-dependent and -independent contributions to theoff-target activity of zinc finger nucleases. Nucleic Acids Res, 2011, 39(1): 381–392.
[10] Sander JD, Ramirez CL, Linder SJ, et al.abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res, 2013, 41(19): e181.
[11] Fine EJ, Cradick TJ, Zhao CL, et al. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res, 2014, 42(6): e42.
[12] Cai YJ, Bak RO, Mikkelsen JG. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife, 2014, 3: e01911.
[13] Miller JC, Holmes MC, Wang JB, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol, 2007, 25(7): 778–785.
[14] Szczepek M, Brondani V, Büchel J, et al. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol, 2007, 25(7): 786–793.
[15] Ramirez CL, Certo MT, Mussolino C, et al. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res, 2012, 40(12): 5560–5568.
[16] Gaj T, Guo J, Kato Y, et al. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods, 2012, 9(8): 805–807.
[17] Deng D, Yan CY, Pan XJ, et al. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science, 2012, 335(6069): 720–723.
[18] Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol, 2013, 14(1): 49–55.
[19] Ding QR, Lee YK, Schaefer EAK, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell, 2013, 12(2): 238–251.
[20] Ke Q, Li WQ, Lai XQ, et al. TALEN-based generation of a cynomolgus monkey disease model for human microcephaly. Cell Res, 2016, 26(9): 1048–1061.
[21] Mussolino C, Alzubi J, Fine EJ, et al. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res, 2014, 42(10): 6762–6773.
[22] Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science, 2009, 326(5959): 1501.
[23] Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science, 2009, 326(5959): 1509–1512.
[24] Doyle EL, Booher NJ, Standage DS, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res, 2012, 40(W1): W117–W122.
[25] Montague TG, Cruz JM, Gagnon JA, et al. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res, 2014, 42(W1): W401–W407.
[26] Labun K, Montague TG, Gagnon JA, et al. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res, 2016, 44(W1): W272–W276.
[27] Guilinger JP, Pattanayak V, Reyon D, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods, 2014, 11(4): 429–435.
[28] Miller JC, Tan SY, Qiao GJ, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol, 2011, 29(2): 143–148.
[29] Kim Y, Kweon J, Kim A, et al. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol, 2013, 31(3): 251–258.
[30] Miller JC, Zhang L, Xia DF, et al. Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat Methods, 2015, 12(5): 465–471.
[31] Lamb BM, Mercer AC, Barbas CF, III. Directed evolution of the TALE N-terminal domain for recognition of all 5′ bases. Nucleic Acids Res, 2013, 41(21): 9779–9785.
[32] Doyle EL, Hummel AW, Demorest ZL, et al. TAL effector specificity for base 0 of the DNA target is altered in a complex, effector- and assay-dependent manner by substitutions for the tryptophan in cryptic repeat-1. PLoS ONE, 2013, 8(12): e82120.
[33] Lin JF, Chen H, Luo L, et al. Creating a monomeric endonuclease TALE-I-SceI with high specificity and low genotoxicity in human cells. Nucleic Acids Res, 2015, 43(2): 1112–1122.
[34] Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol, 2015, 13(11): 722–736.
[35] Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 2012, 337(6096): 816–821.
[36] Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 2013, 154(6): 1380–1389.
[37] Fu YF, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol, 2013, 31(9): 822–826.
[38] Hou ZG, Zhang Y, Propson NE, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from. Proc Natl Acad Sci USA, 2013, 110(39): 15644–15649.
[39] Kuscu C, Arslan S, Singh R, et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol, 2014, 32(7): 677–683.
[40] Tsai SQ, Zheng ZL, Nguyen NT, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol, 2015, 33(2): 187–197.
[41] Ran FA, Cong L, Yan WX, et al.genome editing usingCas9. Nature, 2015, 520(7546): 186–191.
[42] Müller M, Lee CM, Gasiunas G, et al.CRISPR-Cas9 systems enable specific editing of the human genome. Mol Ther, 2016, 24(3): 636–644.
[43] Kim E, Koo T, Park SW, et al.genome editing with a small Cas9 orthologue derived from. Nat Commun, 2017, 8: 14500.
[44] Chuai GH, Wang QL, Liu Q.meets: towards computational CRISPR-based sgRNA design. Trends Biotechnol, 2017, 35(1): 12–21.
[45] Zhang YL, Ge XL, Yang FY, et al. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci Rep, 2014, 4: 5405.
[46] Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature, 2015, 523(7561): 481–485.
[47] Fu YF, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol, 2014, 32(3): 279–284.
[48] Kim S, Kim D, Cho SW, et al. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res, 2014, 24(6): 1012–1019.
[49] Ramakrishna S, Dad ABK, Beloor J, et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res, 2014, 24(6): 1020–1027.
[50] Lin S, Staahl BT, Alla RK, et al. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife, 2014, 3: e04766.
[51] Zuris JA, Thompson DB, Shu YL, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editingand. Nat Biotechnol, 2015, 33(1): 73–80.
[52] Liu J, Gaj T, Yang YF, et al. Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat Protoc, 2015, 10(11): 1842–1859.
[53] Kim K, Park SW, Kim JH, et al. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res, 2017, 27(3): 419–426.
[54] Gao YP, Wu HB, Wang YS, et al. Single Cas9 nickase induced generation of NRAMP1 knockin cattle with reduced off-target effects. Genome Biol, 2017, 18: 13.
[55] Davis KM, Pattanayak V, Thompson DB, et al. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol, 2015, 11(5): 316–318.
[56] Hemphill J, Borchardt EK, Brown K, et al. Optical control of CRISPR/Cas9 gene editing. J Am Chem Soc, 2015, 137(17): 5642–5645.
[57] Nihongaki Y, Kawano F, Nakajima T, et al. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol, 2015, 33(7): 755–760.
[58] Jain PK, Ramanan V, Schepers AG, et al. Development of light-activated CRISPR using guide RNAs with photocleavable protectors. Angew Chem, 2016, 55(40): 12440–12444.
[59] Wright AV, Sternberg SH, Taylor DW, et al. Rational design of a split-Cas9 enzyme complex. Proc Natl Acad Sci USA, 2015, 112(10): 2984–2989.
[60] Truong DJJ, Kühner K, Kühn R, et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res, 2015, 43(13): 6450–6458.
[61] Dow LE, Fisher J, O’Rourke KP, et al. Induciblegenome editing with CRISPR-Cas9. Nat Biotechnol, 2015, 33(4): 390–394.
[62] Oakes BL, Nadler DC, Flamholz A, et al. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol, 2016, 34(6): 646–651.
[63] Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 2016, 529(7587): 490–495.
[64] Slaymaker IM, Gao LY, Zetsche B, et al. Rationally engineered Cas9 nucleases with improved specificity. Science, 2016, 351(6268): 84–88.
[65] Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 toI nuclease improves the specificity of genome modification. Nat Biotechnol, 2014, 32(6): 577–582.
[66] Tsai SQ, Wyvekens N, Khayter C, et al. Dimeric CRISPR RNA-guidedI nucleases for highly specific genome editing. Nat Biotechnol, 2014, 32(6): 569–576.
[67] Wyvekens N, Topkar VV, Khayter C, et al. Dimeric CRISPR RNA-guidedI-dCas9 nucleases directed by truncated gRNAs for highly specific genome editing. Hum Gene Ther, 2015, 26(7): 425–431.
[68] Komor AC, Kim YB, Packer MS, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 2016, 533(7603): 420–424.
[69] Nishida K, Arazoe T, Yachie N, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science, 2016, doi: 10.1126/science.aaf8729. (in Press)
[70] Hess GT, Frésard L, Han K, et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods, 2016, 13(12): 1036–1042.
[71] Kim YB, Komor AC, Levy JM, et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol, 2017, 35(4): 371–376.
[72] Zong Y, Wang YP, Li C, et al. Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat Biotechnol, 2017, 35(5): 438–440.
[73] Kim K, Ryu SM, Kim ST, et al. Highly efficient RNA-guided base editing in mouse embryos. Nat Biotechnol, 2017, 35(5): 435–437.
[74] Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell, 2015, 163(3): 759–771.
[75] Kim Y, Cheong SA, Lee JG, et al. Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol, 2016, 34(8): 808–810.
[76] Hur JK, Kim K, Been KW, et al. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol, 2016, 34(8): 807–808.
[77] Kim H, Kim ST, Ryu J, et al. CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun, 2017, 8: 14406.
[78] Xu RF, Qin RY, Li H, et al. Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol J, 2017, 15(6): 713–717.
[79] Wang MG, Mao YF, Lu YM, et al. Multiplex gene editing in rice using the CRISPR-Cpf1 system. Mol Plant, 2017, 10(7): 1011–1013.
[80] Kim D, Kim J, Hur JK. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol, 2016, 34(8): 863–868.
[81] Kleinstiver BP, Tsai SQ, Prew MS, et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol, 2016, 34(8): 869–874.
[82] Zischewski J, Fischer R, Bortesi L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv, 2017, 35(1): 95–104.
[83] Vouillot L, Thélie A, Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3, 2015, 5(3): 407–415.
[84] Feng ZY, Mao YF, Xu NF, et al. Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in. Proc Natl Acad Sci USA, 2014, 111(12): 4632–4637.
[85] Smith C, Gore A, Yan W, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell, 2014, 15(1): 12–13.
[86] Paix A, Wang YM, Smith HE, et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in. Genetics, 2014, 198(4): 1347–1356.
[87] Ghorbal M, Gorman M, Macpherson CR, et al. Genome editing in the human malaria parasiteusing the CRISPR-Cas9 system. Nat Biotechnol, 2014, 32(8): 819–821.
[88] Veres A, Gosis BS, Ding QR, et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell, 2014, 15(1): 27–30.
[89] Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet, 2009, 10(10): 669–680.
[90] Teytelman L, Thurtle DM, Rine J, et al. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci USA, 2013, 110(46): 18602–18607.
[91] Gabriel R, Lombardo A, Arens A, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol, 2011, 29(9): 816–823.
[92] Wang XL, Wang YB, Wu XW, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol, 2015, 33(2): 175–178.
[93] Osborn MJ, Webber BR, Knipping F, et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther, 2016, 24(3): 570–581.
[94] Crosetto N, Mitra A, Silva MJ, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods, 2013, 10(4): 361–365.
[95] Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol, 2016, 34(2): 184–191.
[96] Chiarle R, Zhang Y, Frock RL, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell, 2011, 147(1): 107–119.
[97] Frock RL, Hu JZ, Meyers RM, et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol, 2015, 33(2): 179–186.
[98] Hu JZ, Meyers RM, Dong JC, et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification–mediated high-throughput genome-wide translocation sequencing. Nat Protoc, 2016, 11(5): 853–871.
[99] Kim D, Bae S, Park J, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Biotechnol, 2015, 12(3): 237–243.
[100] Kim D, Kim S, Kim S, et al. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res, 2016, 26(3): 406–415.
[101] Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. CIRCLE-seq: a highly sensitivescreen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods, 2017, 14(6): 607–614.
[102] Cameron P, Fuller CK, Donohoue PD, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods, 2017, 14(6): 600–606.
(本文责编 陈宏宇)
Genome-editing: focus on the off-target effects
Xiubin He, and Feng Gu
State Key Laboratory of Ophthalmology and Optometry, School of Ophthalmology and Optometry, Wenzhou Medical University, Wenzhou 325000, Zhejiang, China
Breakthroughs of genome-editing in recent years have paved the way to develop new therapeutic strategies. These genome-editing tools mainly include Zinc-finger nucleases (ZFNs), Transcription activator-like effector nucleases (TALENs), and clustered regulatory interspaced short palindromic repeat (CRISPR)/Cas-based RNA-guided DNA endonucleases. However, off-target effects are still the major issue in genome editing, and limit the application in gene therapy. Here, we summarized the cause and compared different detection methods of off-targets.
genome editing, off-target, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), clustered regulatory interspaced short palindromic repeats (CRISPRs)
May 5, 2017;
August 18, 2017
Feng Gu. Tel: +86-577-88831367; E-mail: gufenguw@gmail.com
Supported by:National Basic Research Program of China (973 Program) (No. 2013CB967502), National Natural Science Foundation of China (No. 81201181), Science and Technology Project of Zhejiang Province (No. 2017C37176).
国家重点基础研究发展计划 (973计划) (No. 2013CB967502),国家自然科学基金 (No. 81201181),浙江省科技项目 (No. 2017C37176)资助。
谷峰 博士,温州医科大学研究员,中国遗传学会基因组编辑分会委员,任《遗传》和Scientific Reports 期刊的编委。主要从事新型基因组编辑工具研发、遗传病致病基因发现与相关的基因/干细胞治疗研究。在Cell Stem Cell、Nucleic Acids Research、Molecular Therapy-Nucleic Acids、Human Mutation 等期刊发表多篇文章。

猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
今日农业(2021年11期)2021-08-13
实用临床医药杂志(2021年13期)2021-01-10
中西医结合肝病杂志(2020年2期)2020-10-27
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
中成药(2018年7期)2018-08-04
中国病理生理杂志(2017年2期)2017-01-17
中华胰腺病杂志(2015年5期)2015-12-08
