
在线固相萃取-超高效液相色谱-线性离子阱串联质谱法直接测定水中10种藻类毒素
2017-11-06 03:04:41徐潇颖朱炳祺梁晶晶陈万勤罗金文
分析化学 2017年11期
徐潇颖 刘 柱 朱炳祺 梁晶晶 陈万勤 罗金文
(浙江省食品药品检验研究院, 杭州 310052)
在线固相萃取-超高效液相色谱-线性离子阱串联质谱法直接测定水中10种藻类毒素
徐潇颖 刘 柱 朱炳祺 梁晶晶 陈万勤 罗金文*
(浙江省食品药品检验研究院, 杭州 310052)
建立了全自动在线固相萃取-超高效液相色谱-线性离子阱串联质谱法直接测定水中10种藻类毒素的方法。利用程序实现多次进样,通过在线固相萃取对藻类毒素进行富集,然后切换六通阀,将富集的目标物冲洗至分析柱进行分离后,进入线性离子阱质谱检测。10种藻类毒素在相应的浓度范围内线性关系良好,相关系数均大于0.99,检出限在0.0015~0.0050 μg/L之间,3个浓度水平(0.02、0.10和1.00 μg/L)的加标回收率为83.7%~98.5%。结果表明,在线固相萃取极大简化了前处理过程,线性离子阱串联质谱法提高了痕量藻类毒素测定的灵敏度,增强子离子扫描(EPI)谱库的建立为藻类毒素的确证提供保障。本方法适用于水体中多种藻类毒素的快速确证和定量测定。
藻类毒素; 在线固相萃取; 超高效液相色谱-线性离子阱串联质谱; 水样
2017-05-27收稿; 2017-08-29接受
本文系浙江省食品药品监管系统科技计划项目(No. SP201704)资助
* E-mail: luojw31@163.com
1 引 言
近年来,水体富营养化所导致的水华现象频频出现。水体出现富营养化现象时,藻类大量繁殖,同时代谢产生氰毒素类化合物,主要包括为肝毒素、神经毒素和脂多糖内毒素[1]。肝毒素包括微囊藻毒素(MCs)、节球藻毒素(NOD)和柱孢藻毒素(CYN)[2~4]。其中微囊藻毒素由一个环七肽和特殊的芳香族氨基酸侧链-AD-DA构成,由于环七肽中含有两个可变氨基酸基团,导致其具有多种异构体,迄今为止被报道的微囊藻毒素已达到100多种[5~7]。MC-LR作为一种最常见的微囊藻毒素,世界卫生组织规定其在水中最高允许量为1 μg/L[8],我国制定的相关饮用水标准对其的限量为1.0 μg/L[9]。
藻类毒素在水体中极低的含量大大增加了分析难度。在常规的液相色谱(HPLC)检测方法中,需通过固相萃取(SPE)柱对大体积的样品进行富集后,才能达到HPLC分析的灵敏度,该方法前处理耗费大量时间,难以实现大批量样品的快速分析[10,11],且检出限较高。为简化样品前处理过程,冯桂学等[12]采用全自动在线固相萃取(Online SPE),实现了样品的自动化在线处理和测定,节约时间的同时避免了大量有机试剂的接触;而对于采用HPLC进行测定时存在灵敏度低的问题,研究者多选用液相色谱-串联质谱(LC-MS/MS)代替HPLC[13,14], 如赵起越等[13]采用LC-MS/MS直接进样,对水中藻类毒素进行测定,检出限达到0.007~0.047 μg/L。
本研究采用全自动在线固相萃取-超高效液相色谱(Online SPE -UHPLC)系统结合线性离子阱串联质谱(LIT-MS/MS)进行样品分析。 通过直接进样,在Online SPE-UHPLC系统中[15,16]分别实现连续的自动化在线固相萃取和洗脱分离的过程。离子阱(Qtrap)功能是通过最后一级四极杆切换的切换,在保留传统三重四极杆功能的同时对给定条件的目标离子进行阱集,提高灵敏度[17]。本方法较传统方法极大简化了前处理操作,提高了检测效率,并且降低了检出限。此外,利用增强子离子扫描(EPI)建立的藻毒素二级谱库可为痕量检测提供判定依据,为水体藻类毒素的监管提供科学的依据。
2 实验部分
2.1仪器与试剂
AB Qtrap5500质谱仪(美国AB Sciex公司);Ultimate 3000双三元超高效液相色谱(美国Thermo Fisher公司);Milli-Q超纯水器(美国Millipore公司);XBridge C18Direct Connect HP 色谱柱(30 mm × 2.1 mm, 10 μm),Oasis HLB Direct Connect HP 色谱柱(30 mm × 2.1 mm, 20 μm);Waters Cortecs C18色谱柱(100 mm × 2.1 mm, 2.7 μm)。
甲醇-乙腈(质谱纯,德国Merck公司);甲酸铵、 乙酸铵(分析纯,国药试剂公司);甲酸(质谱纯,美国Sigma Aldrich公司);微囊藻毒素标准品MC-LY、MC-LW、MC-YR、MC-WR、MC-LA、MC-LF、MC-RR、MC-LR(浓度均为10 μg/mL,北京振翔公司);CYN、NOD(纯度均≥95%,加拿大TRC公司)。
2.2混合标准溶液配制
分别称取适量CYN和NOD固体标准品,用甲醇溶解,配制5 μg/mL的标准储备液;分别准确移取适量CYN、NOD标准储备液和其它8种微囊藻毒素液体标准品,用20%(V/V)甲醇溶液稀释配制为1 μg/mL的混合标准使用溶液,于18℃下保存。
2.3在线固相萃取高效液相色谱-串联质谱分析条件
在线固相萃取超高效液相色谱条件:在全自动Online-SPE以及分析系统中均采用甲醇为流动相A,5 mmol/L甲酸铵为流动相B。进样量400 μL(采用变色龙软件连续进样2次实现,每次进样200 μL),上样泵对应在线富集过程流速为0.8 mL/min,分析泵对应目标物在分析柱上的洗脱分离过程流速为0.4 mL/min,柱温35℃,通过六通阀切换来实现在线富集以及洗脱分离过程的连续进行,梯度程序及阀切换时间表见表1。0~2 min,分析泵进行分析柱平衡,上样泵通过连续进样后完成富集过程,比例阀位置于1~2,如图1; 2 min时,六通阀切换至6~1,对富集柱进行反相洗脱,6 min后六通阀切回初始位置,此过程中分析泵通过改变流动相的比例,对进入分析柱的目标物进行分离,最后流入检测器。
表1 在线固相萃取梯度洗脱程序及阀切换时间
Table 1 Gradient elution program and valve-switching time of online solid phase extraction (SPE)
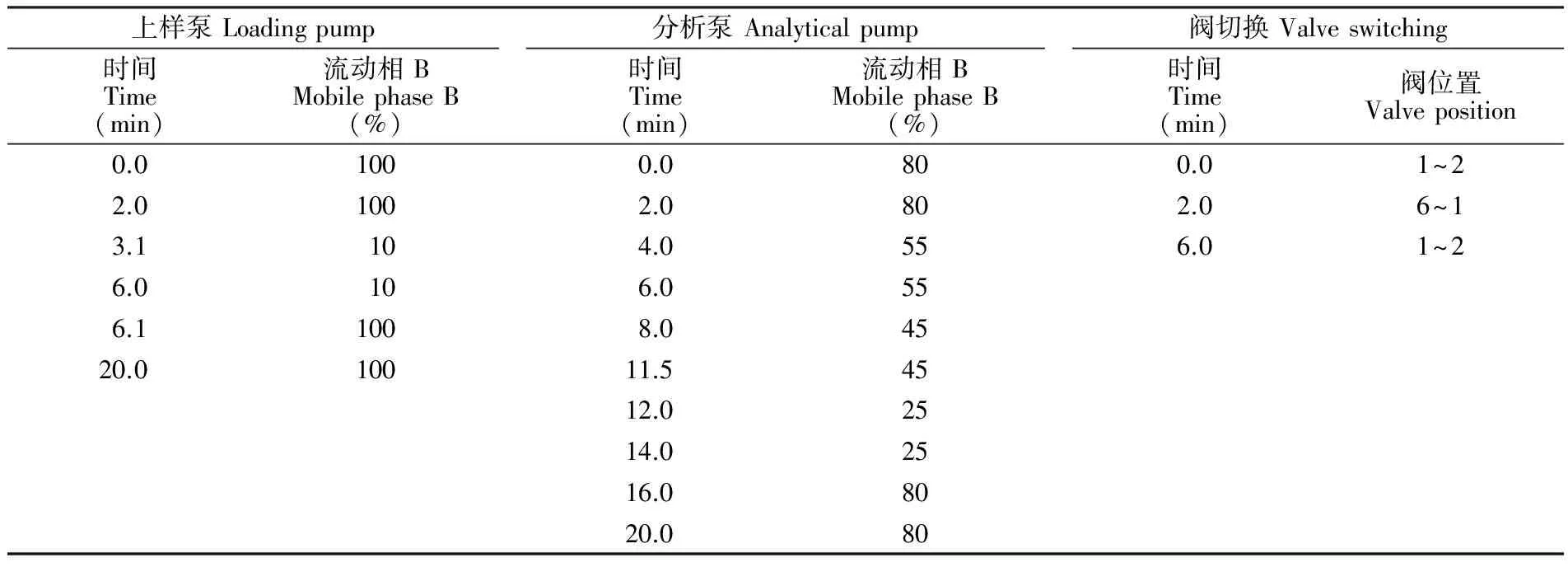
上样泵Loadingpump时间Time(min)流动相BMobilephaseB(%)分析泵Analyticalpump时间Time(min)流动相BMobilephaseB(%)阀切换Valveswitching时间Time(min)阀位置Valveposition0.01000.0800.01~22.01002.0802.06~13.1104.0556.01~26.0106.0556.11008.04520.010011.54512.02514.02516.08020.080
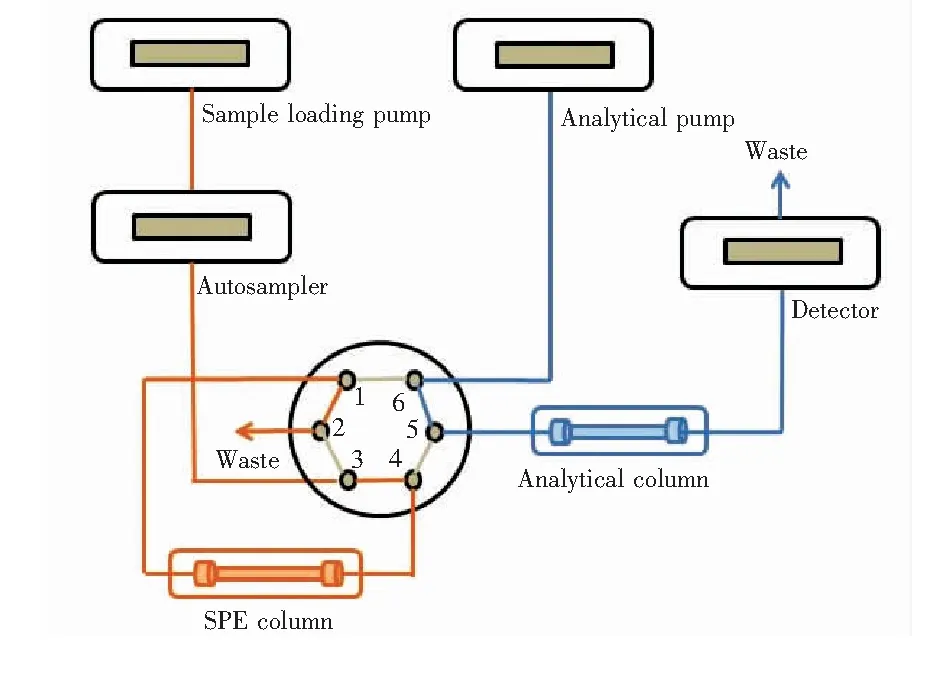
图1 全自动在线固相萃取流路示意图Fig.1 Flow schematic illustration of fully automated SPE
质谱分析条件:电喷雾正离子模式,喷雾电压4.5 kV,雾化器压力(GS1)40 psi,辅助器压力(GS2)40 psi,离子温度450℃,气帘气压力(CUR)35 psi;优化后各目标物的母离子、子离子、碰撞能量和锥孔电压的多反应监测(MRM)模式相关参数见表2。
采用MRM-信息依赖性采集(IDA)-EPI模式检测,建立在线EPI谱库定性分析,IDA参数:启动EPI阈值为5000 CPS,采用动态背景扣除模式;EPI参数:扫描速度10000 Da/s,扫描范围m/z100~1100,采用动态填充阱集时间,且不大于1 ms,EPI碰撞能量:(35 ± 15)eV。
表2 藻类毒素的质谱分析条件
Table 2 ESI-MS/MS parameters for algal toxins analysis
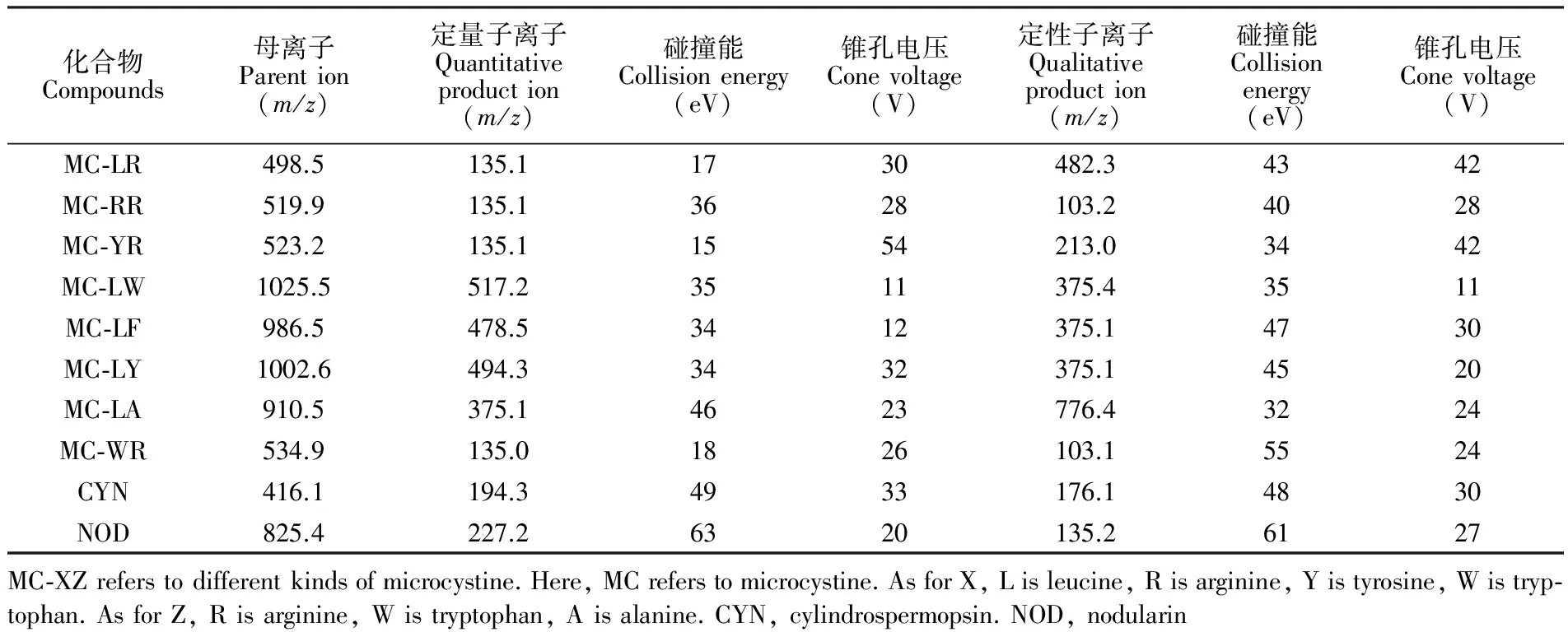
化合物Compounds母离子Parention(m/z)定量子离子Quantitativeproduction(m/z)碰撞能Collisionenergy(eV)锥孔电压Conevoltage(V)定性子离子Qualitativeproduction(m/z)碰撞能Collisionenergy(eV)锥孔电压Conevoltage(V)MC⁃LR498.5135.11730482.34342MC⁃RR519.9135.13628103.24028MC⁃YR523.2135.11554213.03442MC⁃LW1025.5517.23511375.43511MC⁃LF986.5478.53412375.14730MC⁃LY1002.6494.33432375.14520MC⁃LA910.5375.14623776.43224MC⁃WR534.9135.01826103.15524CYN416.1194.34933176.14830NOD825.4227.26320135.26127MC⁃XZreferstodifferentkindsofmicrocystine.Here,MCreferstomicrocystine.AsforX,Lisleucine,Risarginine,Yistyrosine,Wistryp⁃tophan.AsforZ,Risarginine,Wistryptophan,Aisalanine.CYN,cylindrospermopsin.NOD,nodularin
3 结果与讨论
3.1固相萃取色谱柱的选择
在线固相萃取过程中,富集柱的正确选择至关重要。理想的富集柱需要在低有机相条件下实现基质中目标物的有效捕集,而在阀切换启动洗脱程序后,富集柱中的目标物可以迅速被洗脱,并进入分析柱,而不存在因难以洗脱而导致的拖尾等问题。在对微囊藻毒素进行大体积水样离线富集过程中多采用C18和HLB固相萃取柱[18~21],故在本实验中选用HLB和C18富集柱进行比较,结果表明,在最终实验条件下,以100%水相富集2 min时,C18柱和HLB柱对目标物的捕集回收率分别为92.5%~97.8%和95.8%~96.9%,均能满足固相萃取过程对目标化合物的富集要求。在反冲过程中,随着有机相比例逐渐升高,因为HLB trap柱对藻类毒素的保留效果优于C18柱,易造成目标物的色谱峰拖尾,定量积分不准确。故在本实验中选用XBridge C18Direct Connect HP作为富集柱。
3.2流动相选择
实验考虑阀切换后,富集柱中的目标物以及流动相最终进入质谱进行定性与定量检测,所以需选用挥发性较好的流动相,流动相B分别采用0.1%甲酸-水和5 mmol/L甲酸铵代替液相色谱分析藻类毒素时常用的K2PO4(pH 2.5)进行比较分析。结果表明,5 mmol/L甲酸铵所获得的目标物峰形及响应优于0.1%甲酸-水,主要是由于酸性缓冲溶液提高离子强度,有助于改善峰形。在甲醇和乙腈中,流动相A选用甲醇,主要是因为乙腈洗脱能力强,会在反冲过程中使扩散于富集柱中的目标物出现拖尾现象。而在线固相萃取条件中,采用100%水相,10种藻类毒素可在XBridge C18Direct Connect HP 富集柱上得到良好富集,随有机相升高至40%,目标物逐渐被洗脱,流动相中缓冲盐含量以及酸碱度对洗脱效果未造成显著影响。本实验中,为防止富集与分离时流动相的不同对分离过程的影响,在线固相萃取时,液相色谱的流动相与分析过程中的液相流动相一致。
3.3质谱扫描模式的建立
3.3.1MRM条件优化分别利用大气压化学电离源(APCI)、电喷雾离子源(ESI)正、负离子检测模式(ESI+、ESI)对10种藻类毒素进行考察。结果表明,使用APCI时未见信号响应;使用ESI时信号极低,其中MC-LW、MC-WA未见信号;而使用ESI+模式时,10种藻类毒素响应均显著优于ESI模式。文献[22,23]中采用ESI+模式,主要是因为MCs的极性结构使其易于形成正离子,其中MC-LR、MC-YR、MC-RR离子容易结合两个H+,并在m/z498.5[M + 2H]2+、519.9 [M + 2H]2+、523.2 [M + 2H]2+产生较高响应,因此选择上述离子作为母离子,得到的10种藻类毒素的MRM色谱图见图2 。
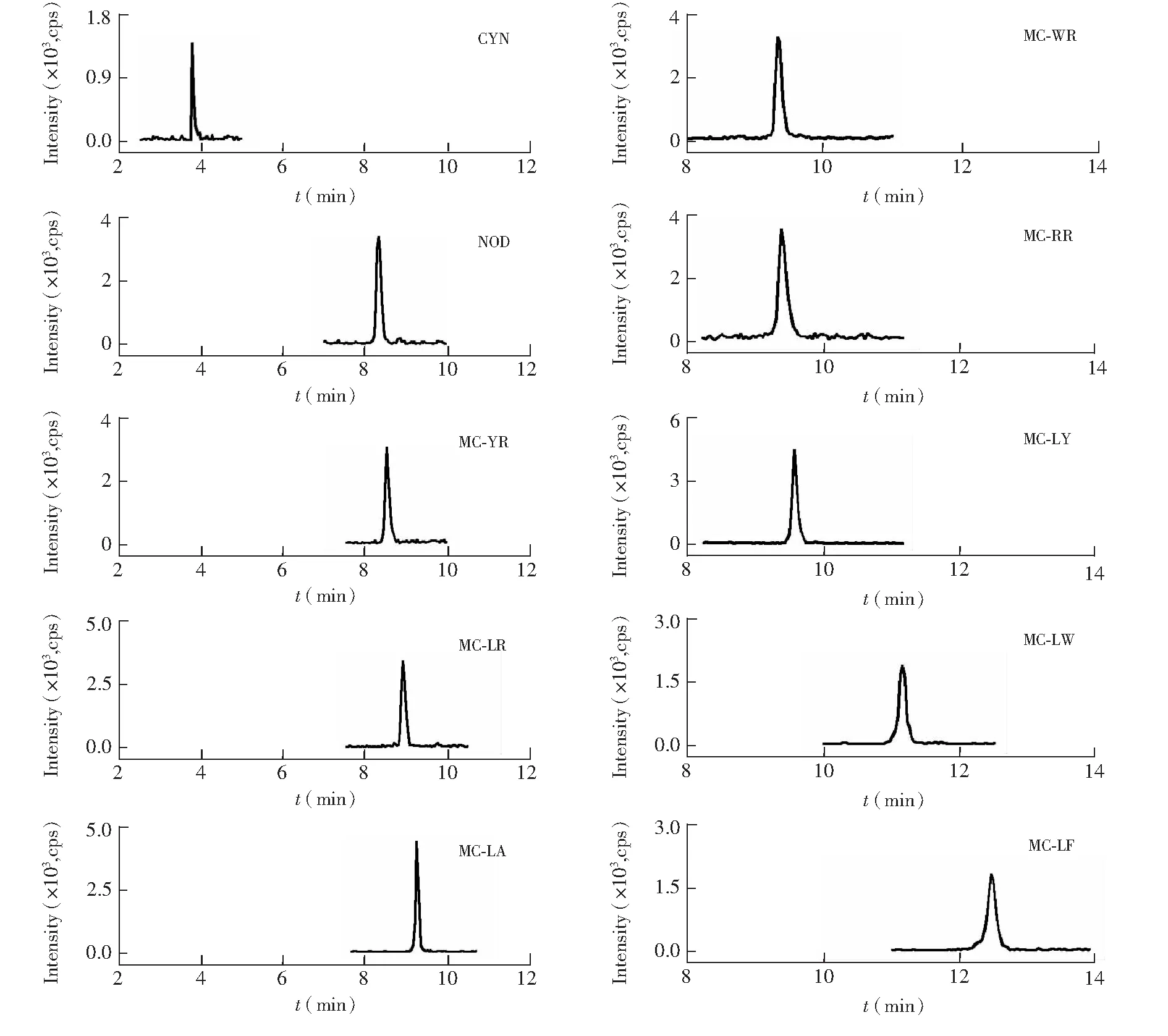
图2 10种藻类毒素的色谱图Fig.2 Chromatograms of 10 kinds of algal toxins
3.3.2MRM-IDA-EPI扫描模式和EPI谱库的建立由于藻类毒素在水体中含量低,采用MRM法进行分析时,质谱绝对响应值较低,目标分析物容易受到基质干扰的影响,定性离子和定量离子丰度比率易超出容许范围,影响定性判定,造成假阳性或假阴性的情况。而Qtrap可将满足MRM模式的目标物的离子进行阱集,经过累积的离子在一定碰撞能下获得更多二级碎片离子,随后进入检测器扫描获得EPI谱图。对增强二级碎片离子的EPI扫描,可以在LC-MS/MS的基础上进一步提高检测灵敏度,通过一次进样可得到用于定量的MRM色谱图和用于定性的增强二级全扫描质谱图,更有利于痕量目标化合物的准确定性分析。
采用优化后的MRM条件进行EPI促发,启动阈值为1000 CPS,选择动态背景扣除模式,扫描速度为10000 Da/s,设置的扫描范围为m/z100~1100 Da,动态阱集时间不大于1 ms,EPI碰撞能量为35 eV,对0.5 ng/mL混合标准溶液进行分析,结果见图3。在该碰撞能下除NOD外均能获得包含各目标物特征碎片的信息,而NOD则需将碰撞能提高至50 eV才能获得足够的信息碎片。将获得的子离子增强二级质谱图编辑谱库,补充对应的目标物名称、相对分子量、CAS号以及化学结构图,在进行样品中痕量藻类毒素测定时,可利用MRM-IDA-EPI扫描模式获取样品中疑似目标物的EPI谱图进行定性谱库检索,特别是在检出限浓度附近进行质谱库检索时,其匹配度均大于85%这一功能是线性离子阱所特有的。
3.4方法学验证
3.4.1标准曲线、检出限及定量限取混合标准使用液, 用初始比例流动相进行逐级稀释,得到浓度在0.005~8.0 μg/L范围内的系列标准溶液,以目标物的峰面积y为纵坐标,质量浓度x(μg/L)为横坐标进行线性拟合,并以色谱峰信噪比(S/N)≥3计算方法检出限(LOD),S/N≥10确定方法定量

限(LOQ),结果见表3,各藻类毒素在线性浓度范围内呈良好线性关系,相关系数>0.99,本方法中各藻类毒素的检出限为0.0015~0.0050 μg/L,远低于现行国家标准规定的0.1 μg/L[9],优于同类文献中的结果[13]。Online SPE-UHPLC-MS/MS-QTRAP方法灵敏度高,可以满足对水中痕量藻毒素检测的要求。
表3 10种藻类毒素的标准曲线、线性范围、相关系数及检出限
Table 3 Linear equations, linear ranges, correlation coefficients and LODs for 10 kinds of algal toxins
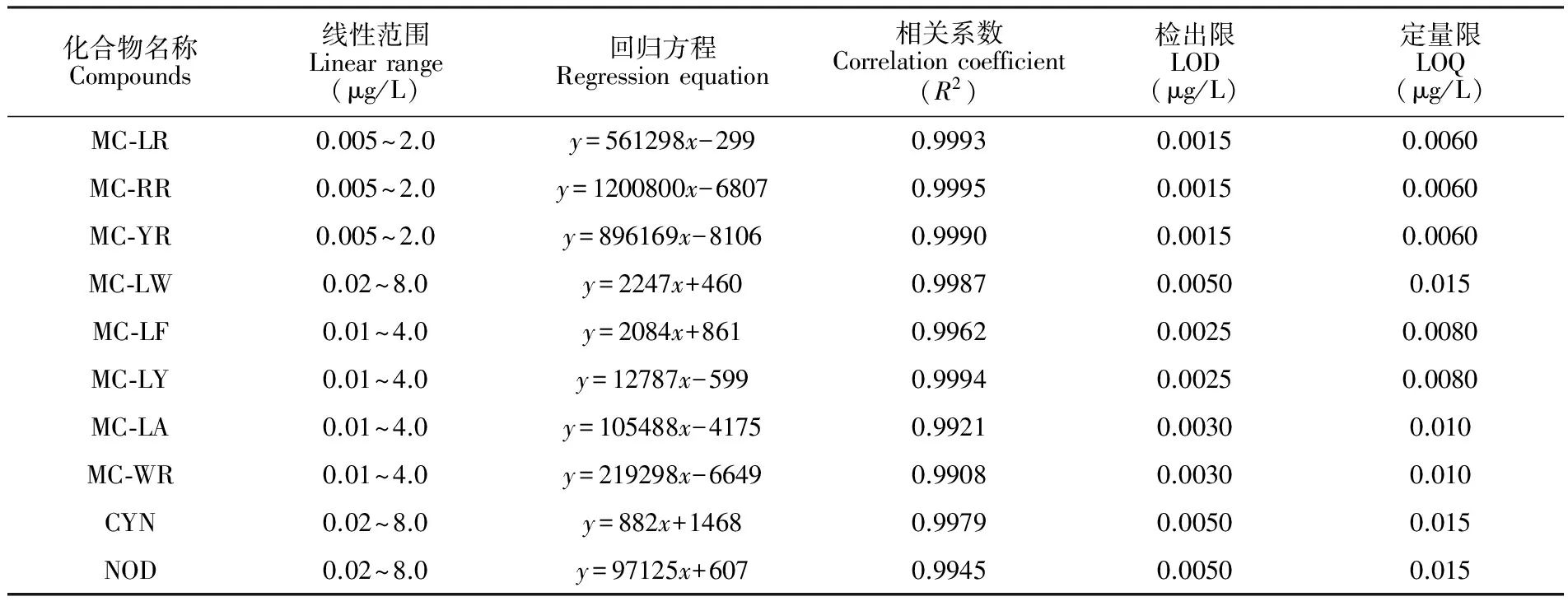
化合物名称Compounds线性范围Linearrange(μg/L)回归方程Regressionequation相关系数Correlationcoefficient(R2)检出限LOD(μg/L)定量限LOQ(μg/L)MC⁃LR0.005~2.0y=561298x-2990.99930.00150.0060MC⁃RR0.005~2.0y=1200800x-68070.99950.00150.0060MC⁃YR0.005~2.0y=896169x-81060.99900.00150.0060MC⁃LW0.02~8.0y=2247x+4600.99870.00500.015MC⁃LF0.01~4.0y=2084x+8610.99620.00250.0080MC⁃LY0.01~4.0y=12787x-5990.99940.00250.0080MC⁃LA0.01~4.0y=105488x-41750.99210.00300.010MC⁃WR0.01~4.0y=219298x-66490.99080.00300.010CYN0.02~8.0y=882x+14680.99790.00500.015NOD0.02~8.0y=97125x+6070.99450.00500.015
3.4.2方法的精密度及准确性在阴性矿泉水水样中加入混合标准溶液,加标水平为0.02、0.10和1.00 μg/L,按本实验优化方法对每个水平样品平行测定6次,计算回收率及相对标准偏差(RSD),结果见表4。在0.02 μg/L的加标水平下,10种藻类毒素的回收率在83.7%~90.2%,相对标准偏差2.8%~4.7%;在0.10 μg/L的加标水平下的回收率在90.5%~97.5%,相对标准偏差2.1%~3.2%;在1.00 μg/L的加标水平下的回收率在92.8%~98.5%,相对标准偏差1.8%~3.2%。结果表明,本方法的准确度和精密度良好,满足水中藻类毒素的测定。
表4 10种藻类毒素的回收率及精密度(n=6)
Table 4 Recoveries and RSDs of 10 kinds of algal toxins(n=6)
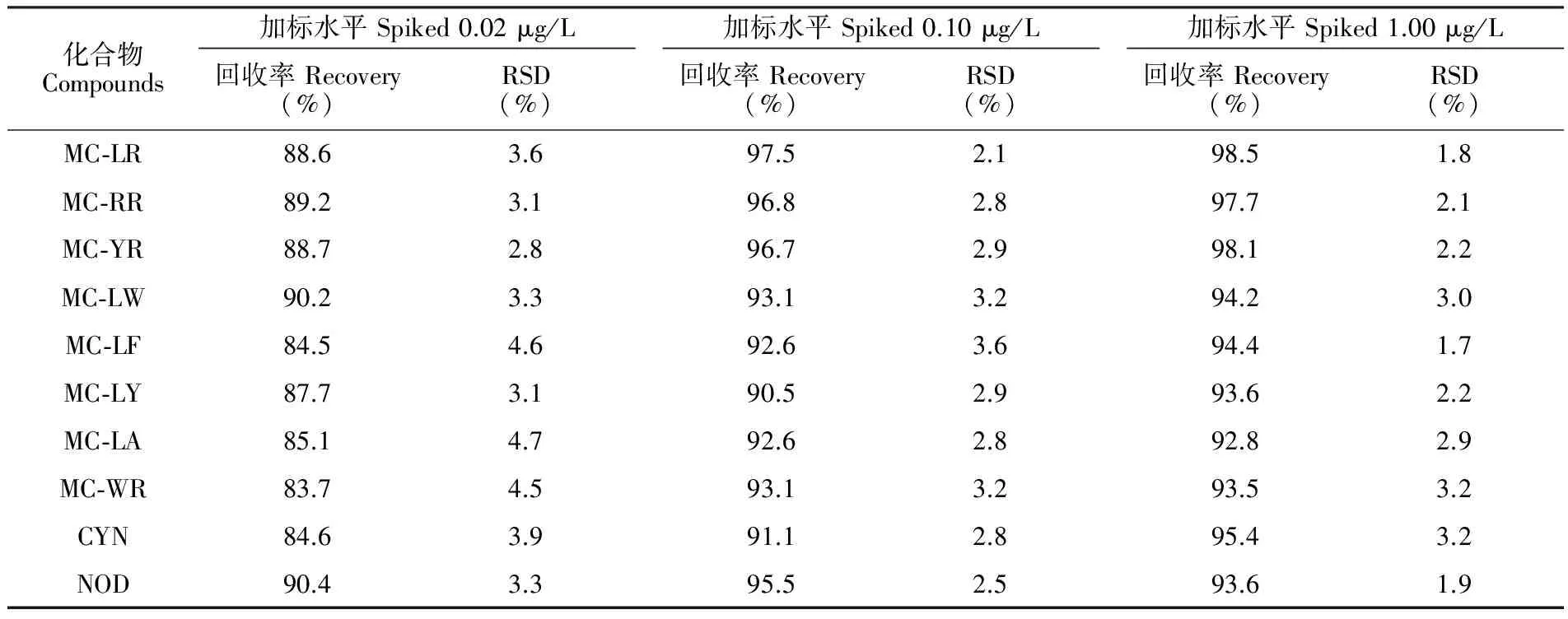
化合物Compounds加标水平Spiked0.02μg/L回收率Recovery(%)RSD(%)加标水平Spiked0.10μg/L回收率Recovery(%)RSD(%)加标水平Spiked1.00μg/L回收率Recovery(%)RSD(%)MC⁃LR88.63.697.52.198.51.8MC⁃RR89.23.196.82.897.72.1MC⁃YR88.72.896.72.998.12.2MC⁃LW90.23.393.13.294.23.0MC⁃LF84.54.692.63.694.41.7MC⁃LY87.73.190.52.993.62.2MC⁃LA85.14.792.62.892.82.9MC⁃WR83.74.593.13.293.53.2CYN84.63.991.12.895.43.2NOD90.43.395.52.593.61.9
3.5实际样品分析
于2017年5月15~17日,对浙江省内包括杭州、湖州、金华地区不同地点的表层水体进行采样,采样时间集中在15~17点,采集29份湖泊水样、 11份河流水样,此外收集20份市售预包装饮用水(包括矿泉水、纯净水、蒸馏水),使用本方法进行测定,结果见表5。 20个品牌市售饮用水中均未检出藻类毒素,根据饮用水中MC-LR、NOD、CYN均不得超过1.0 μg/L安全浓度的规定[8,24],所测定样品均未超标。
表5 10种藻类毒素在表层水样中的测定结果
Table 5 Determination of 10 kinds of algal toxins in surface water samples
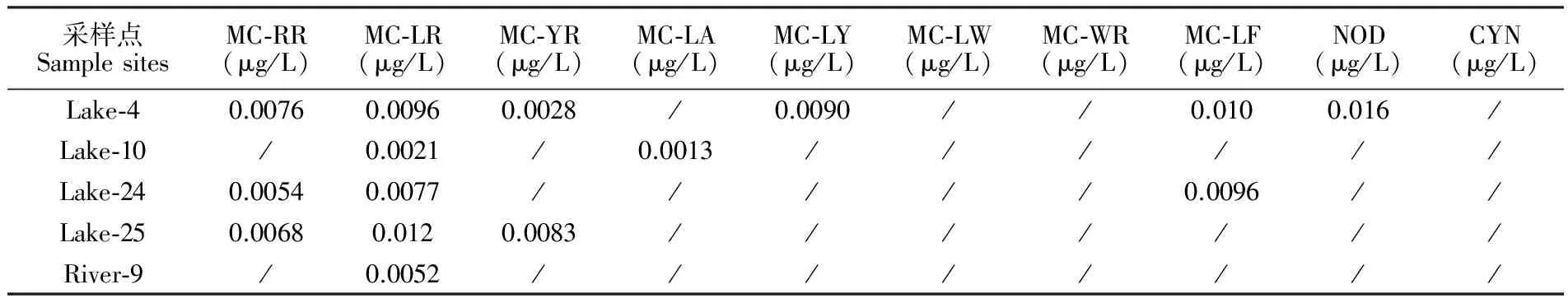
采样点SamplesitesMC⁃RR(μg/L)MC⁃LR(μg/L)MC⁃YR(μg/L)MC⁃LA(μg/L)MC⁃LY(μg/L)MC⁃LW(μg/L)MC⁃WR(μg/L)MC⁃LF(μg/L)NOD(μg/L)CYN(μg/L)Lake⁃40.00760.00960.0028/0.0090//0.0100.016/Lake⁃10/0.0021/0.0013//////Lake⁃240.00540.0077/////0.0096//Lake⁃250.00680.0120.0083///////River⁃9/0.0052////////
4 结 论
本研究建立了Online SPE-UHPLC-MS/MS-QTRAP分析水样中10种藻类毒素的方法。 Online SPE的应用使前处理过程实现了自动化,缩短了前处理的时间,减少了有机试剂使用量;利用UHPLC-MS/MS-QTRAP建立了10种藻类毒素的EPI质谱库,实现了MRM定量检测和在线EPI谱库定性确证的双重功能,很好地解决了传统串联四极杆制谱对低浓度样品定性的问题。Online SPE与UHPLC-MS/MS-QTRAP联用进行水样中痕量藻类毒素测定时,与现有标准和已报道的方法相比,成本更低、分析时间更短、灵敏度更高,检出限可达到0.0015~0.0050 μg/L之间,为藻类毒素的准确定性和定量分析提供了新方法。
1 Bormans M, Lengronne M, Brient L, Duval C.Bull.Environ.Contam.Toxicol.,2014, 92(2): 243-247
2 Chorus I, Bartram J, Falconer I R, Fitzgerald J.Limnol.Oceanogr.,1999, 45(5): 1212-1212
3 Li Xiao-Yu.StudyonMicrocystinsandItsToxicology. Beijing: Science Press,2007
李效宇. 微囊藻毒素及其毒理学研究. 北京: 科学出版社,2007
4 Chen J, Li S C, Guo X C, Xie P, Fan H H, Yu D Z, Zeng C, Chen L.Environ.Toxicol.,2015, (22): 19273-19284
5 Salvador D, Churro C, Valerio E.J.Microbiol.Methods,2016, 123: 4-12
6 Bláhová L, Sejnohová L, Marsálek B, Sejnohová L, Simek Z, Bláha L.Toxicon,2009, 53(5): 519-524
7 Meng G, Sun Y, Fu W, Guo Z, Xu L.Toxicology,2011, 290: 218-229
8 WHO Guidelines for Drinking Watr Quality. Addendum to Volume 2, Health Criteria and Other Supporting Information. 3nd ed. Geneva: World Health Organzaton,2004.
9 GB/T 5749-2006. Standard for Drinking Water Quality. National Standards of the People's Republic of China
生活饮用水卫生标准. 中华人民共和国卫生部. GB/T 5749-2006.
10 Singh S, Srivastava A, 10 Oh H M, Agn C Y, Choi G G, Asthana R K.Toxicon,2012, 60 : 878-894
11 LIANG Li-Li, GONG Ai-Jun, LI Hong-Mei, CAO Yan-Qiu, LI Bao-Qin.ChineseJ.Anal.Chem.,2010, 38(5): 740-742
梁丽丽, 弓爱君, 李红梅, 曹艳秋, 李宝芹. 分析化学,2010, 38(5): 740-742
12 FENG Gui-Xue, LIU Li, WANG Ming-Quan, SUN Shao-Hua, JIA Rui-Bao.ChemicalAnalysisandMeterage,2014, (6): 34-36
冯桂学, 刘 莉, 王明泉, 孙韶华, 贾瑞宝. 化学分析计量,2014, (6): 34-36
13 ZHAO Qi-Yue, ZHAO Hong-Shuai, LIU Bao-Xian, LUO Yang.ChineseJ.Anal.Chem.,2015, 43(4): 594-598
赵起越, 赵红帅, 刘保献, 骆 昉. 分析化学,2015, 43(4): 594-598
14 LAN Ji-Rong, YANG An-Ping, WU Lai-Yan, WANG Song-Bo, JIANG Juan-Juan, WANG Xue-Wu.JournalofInstrmentalAnalysis,2015, 34(6): 664-669
蓝际荣, 杨安平, 吴来燕, 王松波, 蒋娟娟, 王学武. 分析测试学报,2015, 34(6): 664-669
15 Axel M, Ewelina K, Jenny-Maria B, Leif K.Environ.Sci.Pollut.Res.Inter.,2017, 24(9): 8692-8699
16 Pensi D, Nicolo A D, Pinon M, Pisciotta C, Calvo P L, Nonnato A, Romagnoli R, Tandoi F, Perri G D, Avolio A D.J.MassSpectrom.,2017, 52(3): 187-195
17 LIU Zhu, HUA Ying, XU Xiao-Ying, CHEN Wan-Qin, ZHAO Chao-Qun, LUO Jin-Wen.ChineseJ.Anal.Chem.,2016, 44(11): 1728-1734
刘 柱, 华 颖, 徐潇颖, 陈万勤, 赵超群, 罗金文. 分析化学,2016, 44(11): 1728-1734
18 Zhang L, Ping X, Yang Z.Talanta,2014, 62(1): 191-198
19 ZHANG Xiao-Mei, ZHANG Zhu-Qing, HUANG Wen-Peng.ChineseJournalofPublicHealthEngineering,2010, (3): 223-224
张晓梅, 张竹青, 黄文鹏. 中国卫生工程学,2010, (3): 223-224
20 Mekebri A, Blondina G J, Crane D B.J.Chromatogr.A,2009, 1216(15): 3147-3155
21 LIN Shan-Shan, CHEN Meng, LUO Dong-He.The6thNationalConferenceonEnvironmentalChemistry,2011
林珊珊, 陈 猛, 骆和东. 第六届全国环境化学大会暨环境科学仪器与分析仪器展览会,2011
22 YANG Zhen-Yu, ZHOU Yao.JournalofChineseMassSpectrometrySociety,2014, 5(35): 447-453
杨振宇, 周 瑶. 质谱学报,2014, 5(35): 447-453
23 Yao Jin-Ting, Hao Hong-Yuan, Guo Yin, Dong Heng-Tao, Watanbe Kyoko, Huang Tao-Tong, Kawano Shin-ichi, Hashi Yuki. Shimadzu ASMA 2013 WP-033
24 Drinking Water Quality Standards of New Zealand. New Zealand Ministry of Health,2005
SimultaneousDeterminationof10KindsofAlgalToxinsin
WaterSamplesbyOnlineSolidPhaseExtrationCoupled
withUltraHighPerformanceLiquidChromatography-
QuadrupoleLinearIonTrapMassSpectrometry
XU Xiao-Ying, LIU Zhu, ZHU Bing-Qi, LIANG Jing-Jing , CHEN Wan-Qin, LUO Jin-Wen*
(ZhejiangInstituteforFoodandDrugControl,Hangzhou310052,China)
An online solid phase extraction (online-SPE) combined with ultra high performance liquid chromatography-quadrupole linear ion trap mass spectrometry (UHPLC-MS/MS-Qtrap) was established for the simultaneous identification and determination of 10 kinds of algal toxins in water samples. Multiple injections of water samples were controlled by a preset program, and the target analytes were enriched by trap column. The six-way valve was switched subsequently, and the algal toxins in the trap column were back-flushed to the analytical column for separation and analysis. The results showed that the online SPE significantly simplified pretreatment process, and the linear ion trap tandem mass spectrometry improved the sensitivity of the determination. Moreover, the establishment of enhanced product ion (EPI) scan library provided evidence for the confirmation of algal toxins. The 10 kinds of algal toxins showed a good linear relationship with correlation coefficientR2>0.99. The limits of detection (LOD) were 0.0015-0.0050 μg/L. The mean recoveries at three spiked levels of 0.02, 0.1, 1.0 μg/L were from 83.7% to 98.5%. The method was suitable for the rapid confirmation and quantitative determination of various algal toxins in water.
Algal toxins; Online-solid phase extraction; Ultra performance liquid chromatography-quadrupole linear ion trap mass spectrometry; Water
27 May 2017; accepted 29 August 2017)
10.11895/j.issn.0253-3820.170339
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
波谱学杂志(2022年2期)2022-06-14 09:52:02
当代水产(2021年8期)2021-11-04 08:49:00
考试与评价·高二版(2021年3期)2021-09-10 07:22:44
数学物理学报(2020年5期)2020-11-26 06:06:28
军事文摘(2020年20期)2020-11-16 00:31:40
今日农业(2019年10期)2019-01-04 04:28:15
天然产物研究与开发(2018年8期)2018-09-10 05:48:38
环境科技(2016年2期)2016-11-08 12:18:22
公民与法治(2016年14期)2016-05-17 04:15:03
