
分散液液微萃取-气相色谱法快速测定水中15种硝基苯类物质
2017-11-06 03:04:41杜小弟李俊升郭丽萍雷家珩
分析化学 2017年11期
杜小弟 李俊升 郭丽萍 雷家珩
(武汉理工大学化学化工与生命科学学院化学系, 武汉 430070)
分散液液微萃取-气相色谱法快速测定水中15种硝基苯类物质
杜小弟 李俊升 郭丽萍 雷家珩*
(武汉理工大学化学化工与生命科学学院化学系, 武汉 430070)
建立了分散液液微萃取-气相色谱电子捕获检测器测定水中15种硝基苯类物质的方法。筛选出了具有高密度且能够适用于电子捕获检测器的萃取剂。优化了色谱条件,对萃取剂种类及用量、分散剂种类及用量、萃取时间、萃取温度等条件进行了优化。DB-35毛细管柱对15种硝基苯类物质具有最好的分离效果。使用程序升温,初始80℃保持2 min,以5℃/min速率升温至180℃,可以在22 min内完成分离。以100 μL氯苯作为萃取剂、400 μL甲醇作为分散剂,对5.00 mL水样在室温下进行萃取,仅需30 s即可达到萃取平衡,15种目标物的萃取率均可达到90%以上,富集倍数达到45.0~48.8。离心分离,取下层沉积相进行气相色谱测定,使用电子捕获检测器检测,方法的定量限为0.03~0.15 μg/L,线性范围为0.20~50.0 μg/L,相关系数不低于0.998。方法的相对标准偏差在3.3%~8.9%之间,加标回收率在86.0%~103.5%之间。
气相色谱; 分散液液微萃取; 硝基苯类; 饮用水
2017-08-09收稿;2017-09-18接受
本文系国家自然科学基金项目(No. 21476177)资助
* E-mail: yhx2000@263.net
1 引 言
硝基苯类物质是环保部门重点监控的污染物之一。水中硝基苯类物质一般是通过固相萃取[1~6]或液液萃取[4~6]等方法进行分离富集之后,再采用气相色谱法[1~6]或液相色谱法[7,8]进行测定。其中气相色谱法是现行的标准方法[4~6]。液液萃取法较为简便,但溶剂消耗量大,而且萃取之后常还需要进一步蒸发浓缩。固相萃取法的富集效果较好,但仍然存在试样和溶剂用量大的问题,而且耗时很长。固相微萃取法(Solid-phase microextraction, SPME)[9]、单滴萃取法(Single drop microextraction, SDME)[10]、顶空溶剂微萃取法(Headspace solvent microextraction, HSME)[11]也应用于水中硝基苯类物质的分离富集,效果较好但耗时很长。分散液液微萃取法(DLLME)是近年来出现的一种新型萃取方法[12,13],且已被广泛应用[14,15]。通过DLLME法萃取水中硝基苯类物质已有报道[16~21],但一般采用气相色谱的火焰离子化检测器(GC-FID)[16,17,21]或者质谱检测器(GC-MS)[18~20]进行测定,而灵敏度更高的电子捕获检测器(GC-ECD)却很少使用[22]。这是由于DLLME方法需选择高密度萃取剂(通常为卤代烃),以便于离心收集。但多数卤代烃萃取剂的电负性强,不适用于GC-ECD。为了适应GC-ECD,文献[22,23]提出采用十二醇、甲苯等萃取硝基苯类物质。但这些萃取剂的密度都小于水,不能离心收集,需要在低温下进行悬浮固化(Solidification of floating organic-droplets, SFO)[22],或者需要使用特制的萃取容器[23]。这使DLLME方法的简便性大打折扣。
为了同时发挥DLLME法简便快速和GC-ECD法灵敏度高的优点,需要筛选出密度大于水且能够适用于ECD检测器的萃取剂。文献报道中符合上述两个条件的萃取剂仅有二硫化碳[24]和氯苯[25,26],其中用氯苯萃取硝基苯类物质已经有文献进行了尝试[17,19],但涉及到的硝基苯类物质种类较少,且未使用GC-ECD检测。本研究将文献报道的氯苯和二硫化碳,以及本课题组通过实验筛选出的1,4-二氯丁烷、二氯甲烷、1,2-二氯乙烷共5萃取剂进行了比较,建立了简便、快速、灵敏度高的检测水中硝基苯类物质的方法。
2 实验部分
2.1仪器与试剂
GC2010气相色谱仪(日本岛津公司),配有不分流直接进样口(WBI-2010)和电子捕获检测器(ECD-2010); DB-35弹性石英毛细管色谱柱(30 m×0.32 mm×0.25 μm,美国安捷伦公司); 微量注射器(美国SGE公司)。萃取容器为10 mL尖底玻璃离心管(带磨口塞)。
15种硝基苯类标样物质见表1,其中TNT标液(浓度1.01 g/L,甲醇溶剂),购于中国计量科学研究院,其余纯度均不低于99%,购于上海阿拉丁试剂公司; 氯苯、二硫化碳、1,4-二氯丁烷、二氯甲烷、1,2-二氯乙烷(分析纯,上海阿拉丁试剂公司),重蒸3次后使用; 甲醇、乙腈、丙酮(色谱纯,美国Fisher公司)。实验用水为蒸馏水。
待测试样为武汉市内不同居民区采集的自来水,以及分别采自汤逊湖、东湖的湖水。
表1 本方法测定的15种硝基苯类物质
Table 1 15 kinds of nitroaromatic compounds determined by this method
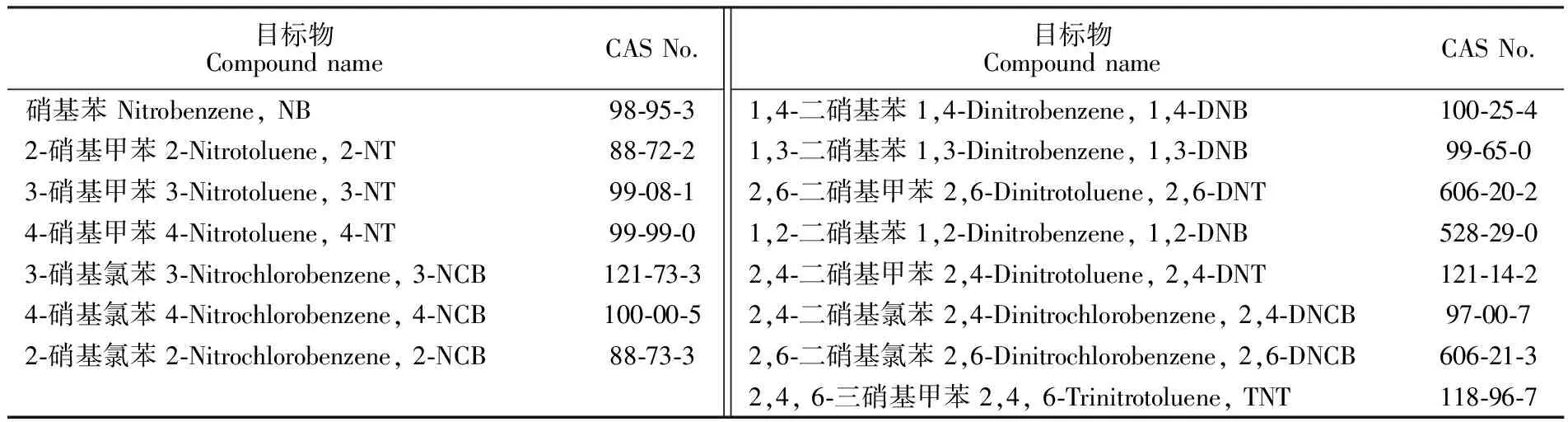
目标物CompoundnameCASNo.硝基苯Nitrobenzene,NB98⁃95⁃32⁃硝基甲苯2⁃Nitrotoluene,2⁃NT88⁃72⁃23⁃硝基甲苯3⁃Nitrotoluene,3⁃NT99⁃08⁃14⁃硝基甲苯4⁃Nitrotoluene,4⁃NT99⁃99⁃03⁃硝基氯苯3⁃Nitrochlorobenzene,3⁃NCB121⁃73⁃34⁃硝基氯苯4⁃Nitrochlorobenzene,4⁃NCB100⁃00⁃52⁃硝基氯苯2⁃Nitrochlorobenzene,2⁃NCB88⁃73⁃3目标物CompoundnameCASNo.1,4⁃二硝基苯1,4⁃Dinitrobenzene,1,4⁃DNB100⁃25⁃41,3⁃二硝基苯1,3⁃Dinitrobenzene,1,3⁃DNB99⁃65⁃02,6⁃二硝基甲苯2,6⁃Dinitrotoluene,2,6⁃DNT606⁃20⁃21,2⁃二硝基苯1,2⁃Dinitrobenzene,1,2⁃DNB528⁃29⁃02,4⁃二硝基甲苯2,4⁃Dinitrotoluene,2,4⁃DNT121⁃14⁃22,4⁃二硝基氯苯2,4⁃Dinitrochlorobenzene,2,4⁃DNCB97⁃00⁃72,6⁃二硝基氯苯2,6⁃Dinitrochlorobenzene,2,6⁃DNCB606⁃21⁃32,4,6⁃三硝基甲苯2,4,6⁃Trinitrotoluene,TNT118⁃96⁃7
2.2实验方法
标准溶液配制:除TNT外,其它标样分别用甲醇配制成1.00 g/L的单标储备液。使用时稀释成所需浓度的混合工作标液。
水样的预处理:水样经0.45 μm尼龙滤膜过滤,移取5.00 mL于尖底玻璃离心管中,用微量注射器将100 μL氯苯(萃取剂)与400 μL甲醇(分散剂)的混合液迅速注入到水样中,加塞轻摇约30 s,得到均匀乳状液。以6000 r/min离心2 min破乳。弃去上层水相,吸取下层沉积相进行色谱分析。
色谱条件:使用DB-35色谱柱,高纯氢气(99.999%)为载气,恒线速度控制(65 cm/s)。程序升温,初始80℃保持2 min,以5℃/min速率升温至180℃,保持5 min。ECD检测器温度220℃,尾吹气为高纯氮气(99.999%),流速40 mL/min。WBI不分流直接进样口,温度200℃,进样1.00 μL。
萃取率(Extraction recovery, ER)和富集倍率(Enrichment factor, EF)按下列公式计算:
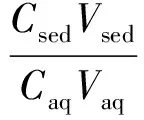
其中,C0为水相中目标物的浓度,模拟样中C0为已知;Csed为沉积相中目标物的浓度,通过气相色谱外标法测得;Vaq为水样体积;Vsed为沉积相体积,由于1 mL水中仅能溶解0.4 μL氯苯,因此近似认为沉积相体积与加入的萃取剂体积相等。
2.3分析方法全程质量控制
以纯水代替实际水样进行空白实验,所测得空白值小于方法检出限,因此测定中未进行空白扣除。将标准储备液用甲醇稀释成混合标样,并添加到水样中进行加标回收实验,测定萃取过程的萃取率和方法的加标回收率。以现行国标方法[5]作为对照方法,采用固相萃取-气相色谱法(SPE-GC-ECD)对相同的水样和加标水样进行测定。
3 结果与讨论
3.1色谱条件优化
3.1.1色谱柱的选择由于目标物均为芳烃衍生物,根据相似相容原则,选择含苯基的聚硅氧烷固定相进行分离。实验结果表明, DB-5和DB-35毛细管柱均可有效分离15种硝基苯类物质,但DB-35的选择性更好,能够获得更高的分离度。因此本研究选择DB-35毛细管柱,15种硝基苯类物质标样的色谱图见图1。
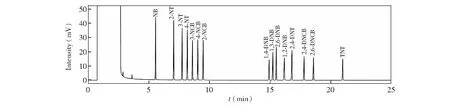
图1 DB-35毛细管色谱柱分离15种硝基苯类物质的色谱图,色谱条件如2.2节所述Fig.1 Chromatograms of 15 nitroaromatic compounds using capillary columns with DB-35 stationary phase, Chromatographic conditions are as described in Section 2.2
3.1.2柱温的优化为了提高灵敏度,本方法采用不分流进样。为了避免进样产生的峰展宽,必须使用较低的初始柱温,使溶剂和目标物在柱头冷凝。分别以60℃、80℃、100℃、120℃为初始柱温进行程序升温,结果表明,初温80℃时可以获得尖锐的峰型,且溶剂峰完全不拖尾,分离时间也较短。
3.1.3检测器温度的优化由于ECD的响应信号具有温度敏感性,本研究考察了检测器温度在220℃~280℃范围内变化时各目标物响应信号的变化。结果表明,各目标物的峰高随检测器温度变化不明显,但溶剂峰的强度随检测器温度的降低而减弱。因此选择220℃作为ECD检测器的温度对减弱溶剂峰的干扰比较有利。
3.2萃取条件的优化
3.2.1萃取剂的种类和用量实验比较了二硫化碳、氯苯和1,4-二氯丁烷、二氯甲烷, 2-二氯乙烷共5种萃取剂。结果表明,二氯甲烷由于在水中溶解度很大而无法形成沉积相,1,2-二氯乙烷也存在类似问题,能够收集到的沉积相太少,难以进行后续实验。二硫化碳、氯苯和1,4-二氯丁烷3种萃取剂可以顺利进行萃取,这3种萃取剂对15种硝基苯类物质的萃取率见图2。二硫化碳对所有目标物的萃取率都较低,1,4-二氯丁烷和氯苯的萃取效果较好。氯苯由于具有与目标物类似的苯环结构,因此具有较好的相容性,从而对大部分目标物都有较高的萃取率。1,4-二氯丁烷对NB、4-NCB、2,4-DNCB这3个目标物的萃取率略高于氯苯,但对其它12种目标物的萃取率明显低于氯苯。综合考虑,选择氯苯作为萃取剂效果较好。

图2 不同萃取剂对15种硝基苯类物质的萃取率的影响,萃取条件如2.2节所述Fig.2 Effect of different extraction solvents on extraction recovery of 15 nitroaromatic compoundsExtraction conditions are as described in Section 2.2
进一步考察了萃取剂用量对萃取效果的影响, 分别使用50、100、150和200 μL氯苯进行萃取(图3),结果表明,使用50 μL氯苯不能完全萃取目标物,特别是NB、1,4-DNB、1,3-DNB、1,2-DNB这4种目标物的萃取率较低,仅约为80%。使用100 μL氯苯萃取时,各目标物的萃取率明显提高,均达到90%以上。进一步提高氯苯用量,萃取率仍可提高。但考虑到富集倍数与萃取剂用量成反比,萃取剂用量过多会导致富集倍数显著降低,为了兼顾富集倍率和萃取率,萃取剂氯苯的最佳用量选择100 μL。
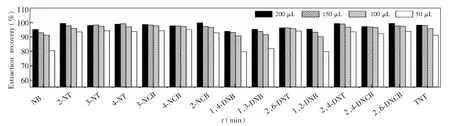
图3 萃取剂(氯苯)用量对15种硝基苯类物质的萃取率的影响,萃取条件如2.2节所述Fig.3 Influence of extraction solvent (chlorobenzene) volume on extraction recovery of 15 kinds of nitroaromatic compoundsExtraction conditions are as described in Section 2.2
3.2.2分散剂的种类和用量DLLME方法中的分散剂需与水和萃取剂都能完全混溶。本研究选择常用的甲醇、乙腈、丙酮3种分散剂进行了对比。3种分散剂在用量相同时对各目标物萃取率的影响见图4。使用丙酮作分散剂时,所有目标物的萃取率都较低。对于硝基甲苯类物质和硝基氯苯类物质,使用甲醇或乙腈作分散剂都能获得较高的萃取率,无明显差异。但对于硝基苯和3种二硝基苯异构体,使用乙腈为分散剂时萃取率明显较低。这可能是由于硝基苯和二硝基苯极性较强且在乙腈溶液中溶解度更大。综合考虑,选择甲醇作为分散剂对目标物进行萃取。
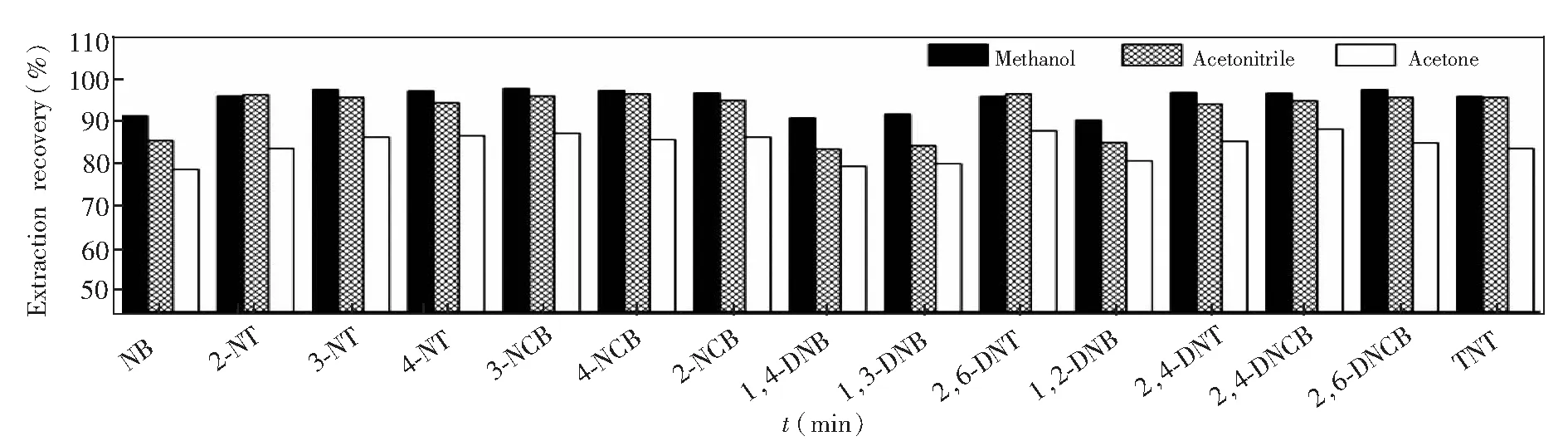
图4 不同分散剂对15种硝基苯类物质的萃取率的影响,萃取条件如2.2节所述Fig.4 Influence of different disperser solvents on extraction recovery of 15 kinds of nitroaromatic compoundsExtraction conditions are described in Section 2.2.
由于DLLME方法中分散剂的用量一般在0.2~1.0 mL左右[15],本研究分别以200、400和600 L甲醇作为分散剂进行萃取。结果表明,200 μL甲醇不能形成稳定的乳状分散体系,必须靠手动振荡进行萃取。使用400和600 μL甲醇作为分散剂时效果较好,都能形成均一、稳定的乳状液,各目标物的萃取率无显著差异。考虑到甲醇用量增加会使硝基苯类物质在水相中溶解度增大,因此本方法选择分散剂的用量为400 μL。
3.2.3萃取的时间和温度根据文献报道,使用DLLME方法可以在很短时间内达到萃取平衡[12,13]。分别在萃取时间为0.5、5和10 min的条件下进行实验,结果表明,各目标物的萃取率基本一致。因此本方法选择萃取时间为0.5 min。
温度变化会改变目标物在两相中的分配系数,对萃取效果会产生一定影响。但一般室内环境温度变化不会太大。分别在15℃、25℃、30℃的水浴条件下进行萃取,发现各目标物的萃取率无显著变化。因此萃取在室温条件下进行。
3.2.4水相的盐浓度和酸度一般认为,通过盐析作用可以提高有机物的萃取率。考察了水样中添加NaCl(0.20~1.00 mol/L)对各种目标物萃取率的影响,结果表明,盐浓度增加时萃取率变化小于3%。这可能是因为硝基苯类物质极性较强,受盐析作用不明显。因此本方法在萃取时不添加无机盐。
实际水样的pH值一般变化范围不大,通常在弱酸性到弱碱性范围,部分水样在采集时为了方便保存而添加HCl酸化。实验采用HCl和NaOH将水样分别调至pH=2.0、4.0、7.0、10.0进行萃取,结果表明,pH值变化对萃取率的影响很小。因此采集的水样可直接进行萃取,不需调节pH值。
3.2.5沉积相的分离由于萃取剂氯苯的密度大于水,以6000 r/min离心2 min可使有机相完全沉积到离心管底部,水相澄清透明。由于萃取过程已经达到平衡,有机相浓度是均一的,在分离不完全的情况下也可以获得一致的测定结果。由于水在氯苯中的溶解度很低,有机相含水很少,无需干燥,可直接进行色谱测定[17,19,25,26]。
3.3方法的分析性能
将混合标液用纯水稀释成不同浓度范围的模拟样品,按2.2节所述条件进行萃取和测定,以各目标物的峰面积对其浓度进行线性回归(表2)。各目标物在0.200~50.0 μg/L范围内均有很好的线性响应,相关系数大于0.998。根据最低浓度标样计算信噪比(S/N),以S/N=3、S/N=10求得方法的检出限和定量限(表2)。由于本方法通过萃取获得了接近50倍的富集因子,因此灵敏度较高,定量限为0.03~0.15 μg/L,完全可以满足地表水中硝基苯类物质检测的要求。
表2 15种硝基苯类物质的线性范围、检出限、定量限和富集因子
Table 2 Linear range, limit of detection (LOD), limit of quantification (LOQ), and enrichment factor of 15 kinds of nitroaromatic compounds
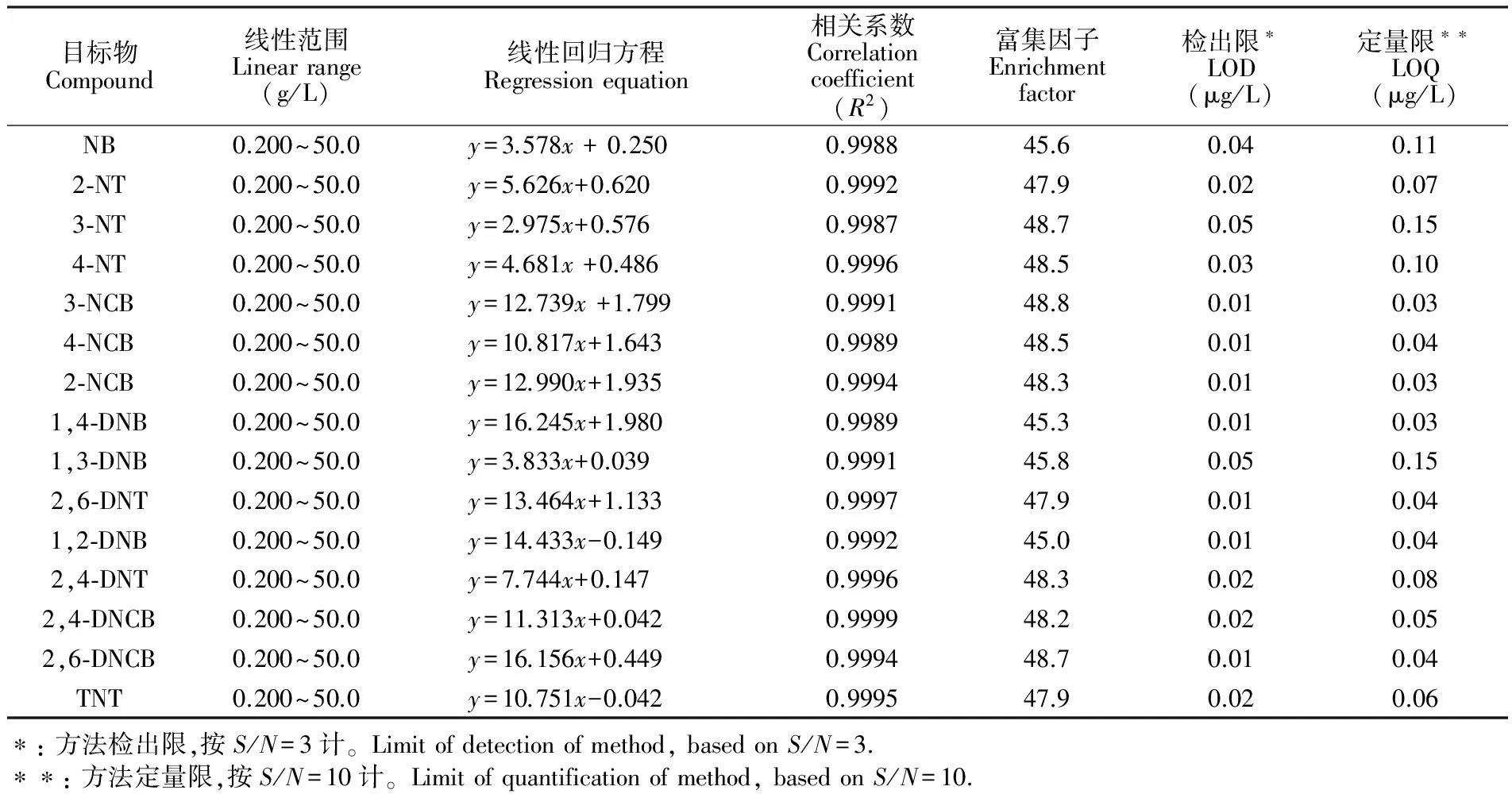
目标物Compound线性范围Linearrange(g/L)线性回归方程Regressionequation相关系数Correlationcoefficient(R2)富集因子Enrichmentfactor检出限∗LOD(μg/L)定量限∗∗LOQ(μg/L)NB0.200~50.0y=3.578x+0.2500.998845.60.040.112⁃NT0.200~50.0y=5.626x+0.6200.999247.90.020.073⁃NT0.200~50.0y=2.975x+0.5760.998748.70.050.154⁃NT0.200~50.0y=4.681x+0.4860.999648.50.030.103⁃NCB0.200~50.0y=12.739x+1.7990.999148.80.010.034⁃NCB0.200~50.0y=10.817x+1.6430.998948.50.010.042⁃NCB0.200~50.0y=12.990x+1.9350.999448.30.010.031,4⁃DNB0.200~50.0y=16.245x+1.9800.998945.30.010.031,3⁃DNB0.200~50.0y=3.833x+0.0390.999145.80.050.152,6⁃DNT0.200~50.0y=13.464x+1.1330.999747.90.010.041,2⁃DNB0.200~50.0y=14.433x-0.1490.999245.00.010.042,4⁃DNT0.200~50.0y=7.744x+0.1470.999648.30.020.082,4⁃DNCB0.200~50.0y=11.313x+0.0420.999948.20.020.052,6⁃DNCB0.200~50.0y=16.156x+0.4490.999448.70.010.04TNT0.200~50.0y=10.751x-0.0420.999547.90.020.06∗:方法检出限,按S/N=3计。Limitofdetectionofmethod,basedonS/N=3.∗∗:方法定量限,按S/N=10计。Limitofquantificationofmethod,basedonS/N=10.
3.4实际样品分析
测定了3个饮用水试样和两个湖水试样,仅有一个湖水试样检出3种硝基苯化合物。有检出试样的色谱图见图5A,经过萃取处理后,试样基体净化效果较好,干扰峰较少且能够有效分离。以该试样为基体,进行了0.200、2.00和20.0 μg/L共3个水平的加标实验,其中加标0.200 μg/L试样的色谱图见图5B。加标试样的测定结果见表3,以现行国标方法作为对照方法的测定结果也列于表3,可以看出,两种方法的结果具有较好的一致性。本方法的相对标准偏差(RSD)和加标回收率见表4。在接近方法的定量限的加标水平(0.200 μg/L)下,方法的相对标准偏差在3.3%~8.9%之间,加标回收率为86.0%~103.5%,可满足痕量分析的要求。在中、高浓度水平(2.00和20.0 μg/L)下,方法的相对标准偏差均不超过5%,加标回收率在94.5%~101.5%之间。
进一步将本方法与文献报道的测定水中硝基苯类物质的方法进行对比(表5)可知,本方法灵敏度高、简便、快速。与现行国标方法相比,样品处理时间由2 h缩短至约3 min,处理效率显著提高。另外,本方法的试样和萃取剂用量少,仅为国标方法的1%,节约了试剂,减少了污染。
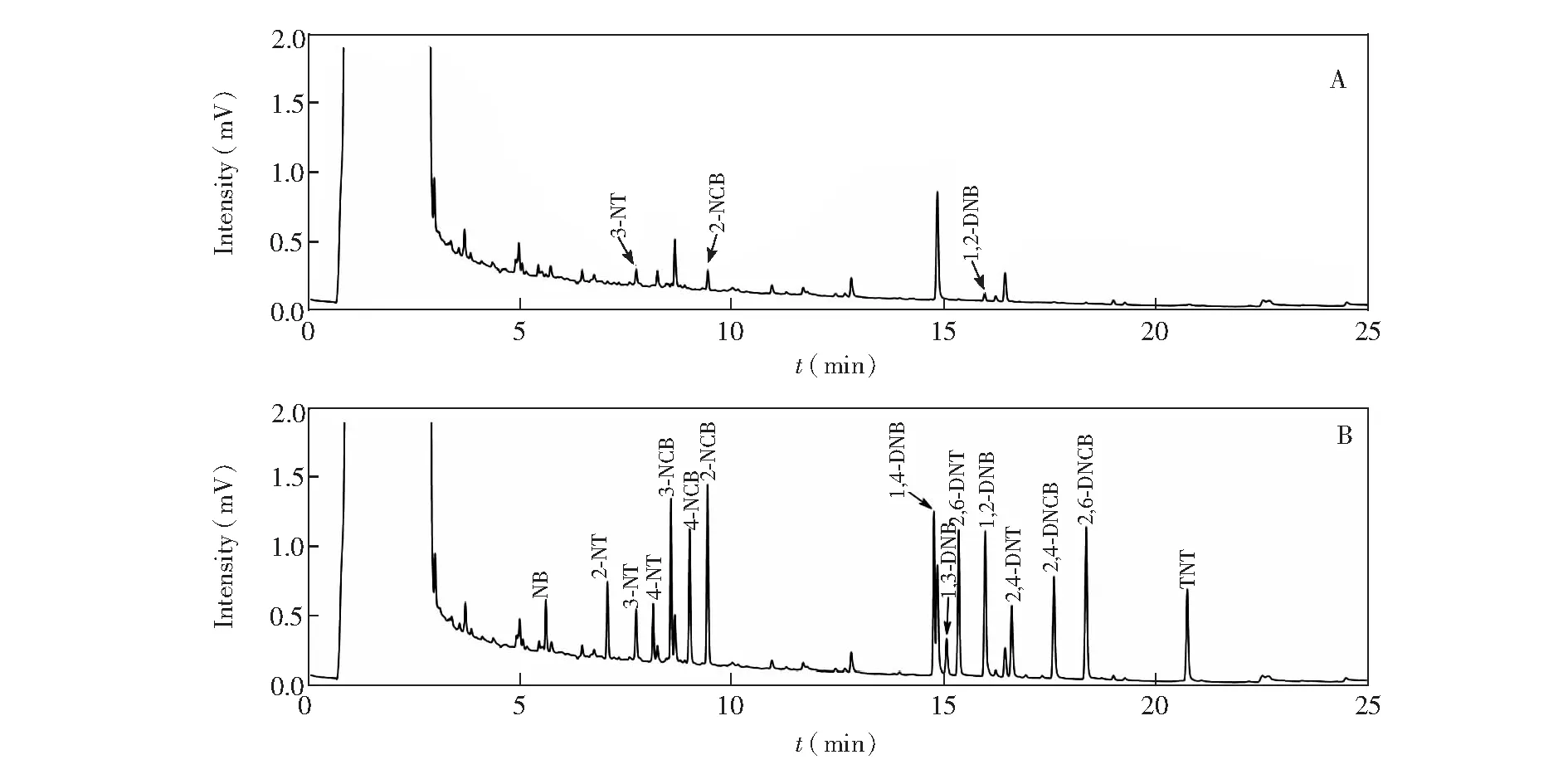
图5 实际水样与加标水样的色谱图Fig.5 Chromatograms of real water sample and spiked water sample obtained by DLLME-GC-ECD methdA:实际水样; B:加标水样,每种目标物的加标水平为0.200 μg/L。色谱条件见2.2节所述A: Real water sample; B: spiked water samples at the concentration level of 0.200 μg/L for each nitroaromatic compound. Chromatographic conditions are as described in Section 2.2
表3 本方法与国标方法测定结果的比较(n=7)
Table 3 Comparison of the results by the proposed method and national standard method (n=7)
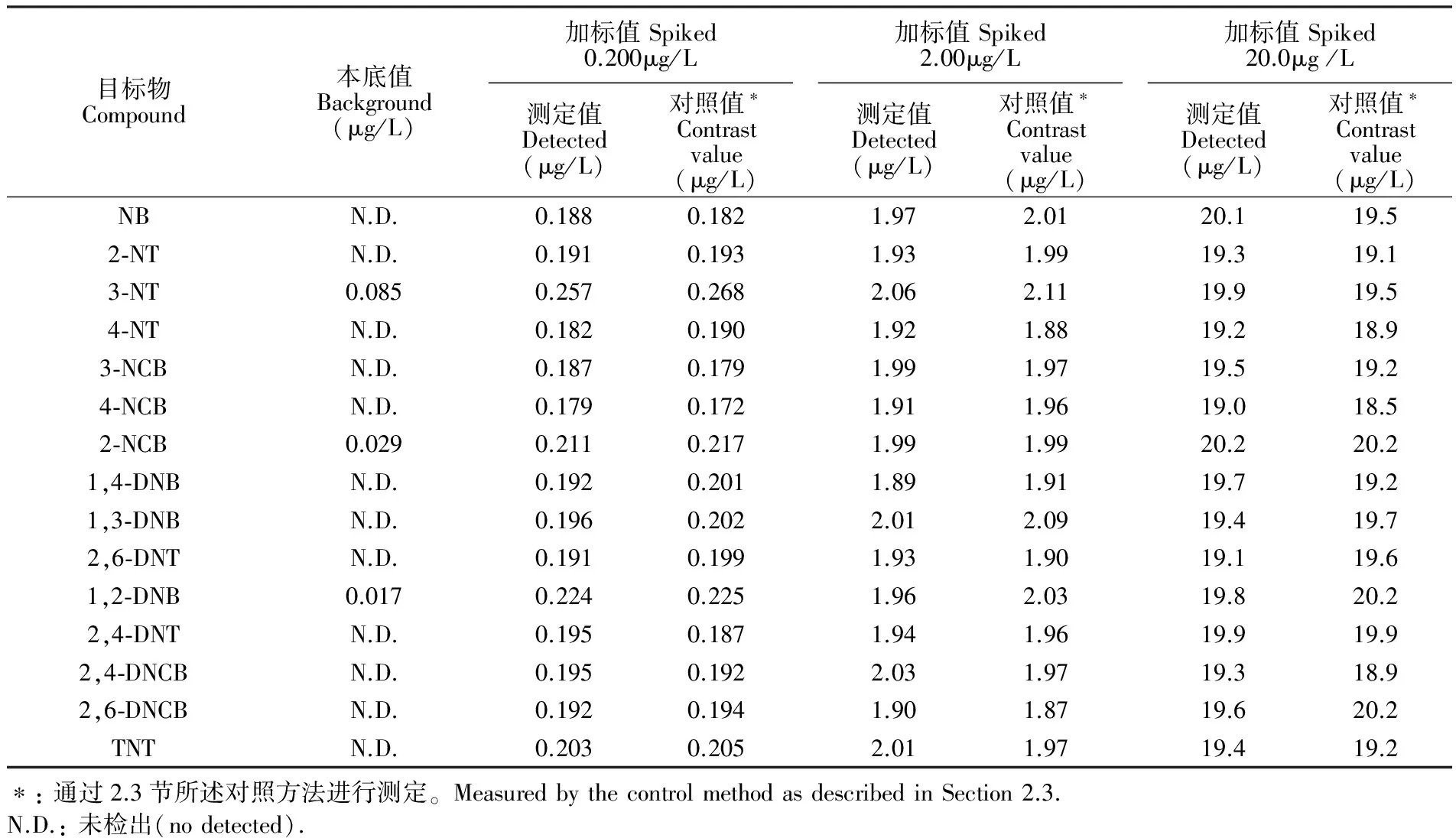
目标物Compound本底值Background(μg/L)加标值Spiked0.200μg/L测定值Detected(μg/L)对照值∗Contrastvalue(μg/L)加标值Spiked2.00μg/L测定值Detected(μg/L)对照值∗Contrastvalue(μg/L)加标值Spiked20.0μg/L测定值Detected(μg/L)对照值∗Contrastvalue(μg/L)NBN.D.0.1880.1821.972.0120.119.52⁃NTN.D.0.1910.1931.931.9919.319.13⁃NT0.0850.2570.2682.062.1119.919.54⁃NTN.D.0.1820.1901.921.8819.218.93⁃NCBN.D.0.1870.1791.991.9719.519.24⁃NCBN.D.0.1790.1721.911.9619.018.52⁃NCB0.0290.2110.2171.991.9920.220.21,4⁃DNBN.D.0.1920.2011.891.9119.719.21,3⁃DNBN.D.0.1960.2022.012.0919.419.72,6⁃DNTN.D.0.1910.1991.931.9019.119.61,2⁃DNB0.0170.2240.2251.962.0319.820.22,4⁃DNTN.D.0.1950.1871.941.9619.919.92,4⁃DNCBN.D.0.1950.1922.031.9719.318.92,6⁃DNCBN.D.0.1920.1941.901.8719.620.2TNTN.D.0.2030.2052.011.9719.419.2∗:通过2.3节所述对照方法进行测定。MeasuredbythecontrolmethodasdescribedinSection2.3.N.D.:未检出(nodetected).
4 结 论
通过DLLME法分离富集水中的15种硝基苯类物质, 氯苯和1,4-二氯丁烷均可作为萃取剂,但氯苯的萃取效果更好,萃取率不低于90%,富集因子接近50。萃取液经DB-35毛细管柱分离,用ECD定量检测,定量限达到0.03~0.15 μg/L。本方法在处理速度上显著优于文献方法,样品和试剂的用量也更少。
表4 方法的精密度及加标回收率(n=7)
Table 4 Precision and recovery results of this method (n=7)
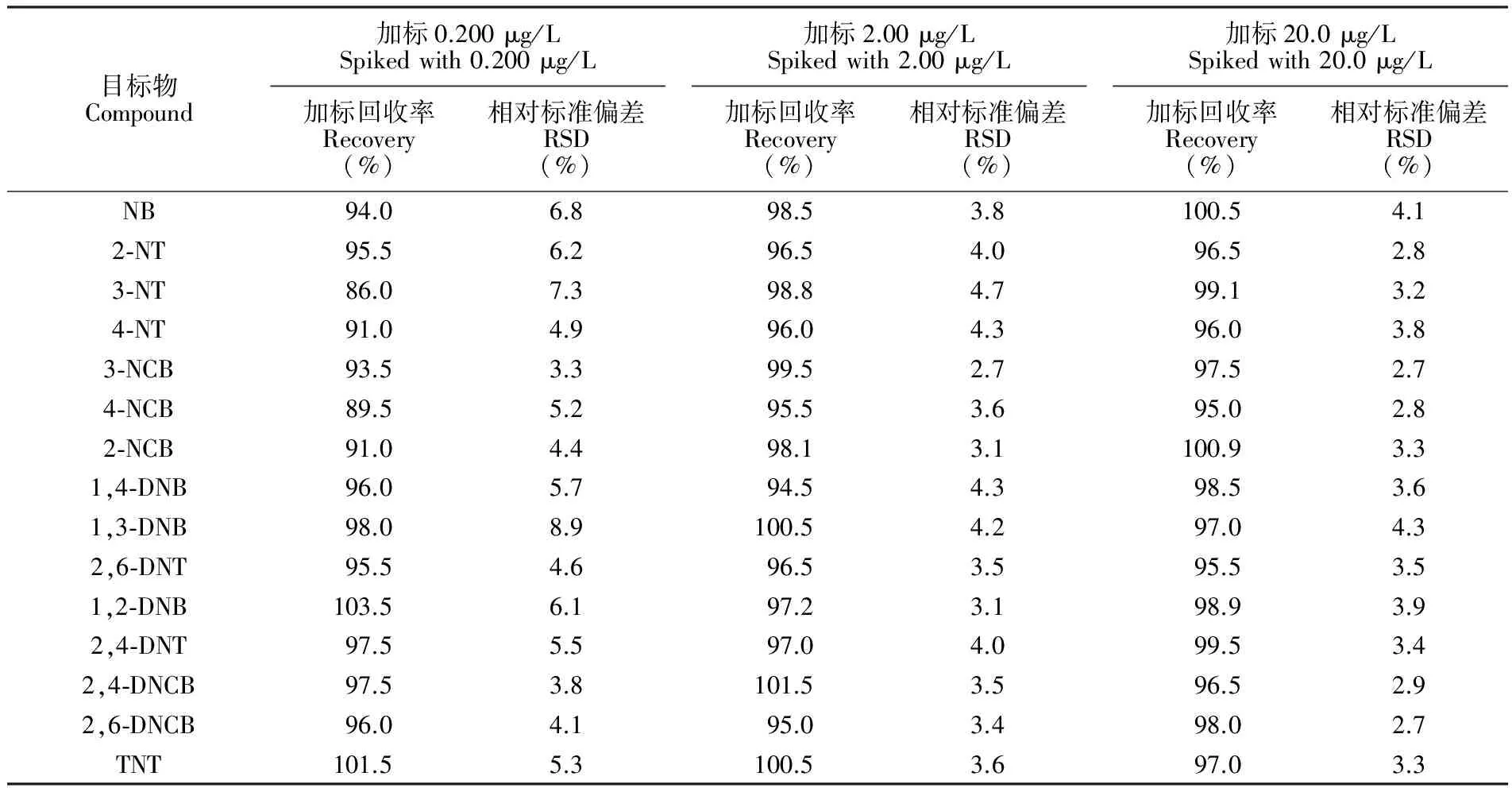
目标物Compound加标0.200μg/LSpikedwith0.200μg/L加标回收率Recovery(%)相对标准偏差RSD(%)加标2.00μg/LSpikedwith2.00μg/L加标回收率Recovery(%)相对标准偏差RSD(%)加标20.0μg/LSpikedwith20.0μg/L加标回收率Recovery(%)相对标准偏差RSD(%)NB94.06.898.53.8100.54.12⁃NT95.56.296.54.096.52.83⁃NT86.07.398.84.799.13.24⁃NT91.04.996.04.396.03.83⁃NCB93.53.399.52.797.52.74⁃NCB89.55.295.53.695.02.82⁃NCB91.04.498.13.1100.93.31,4⁃DNB96.05.794.54.398.53.61,3⁃DNB98.08.9100.54.297.04.32,6⁃DNT95.54.696.53.595.53.51,2⁃DNB103.56.197.23.198.93.92,4⁃DNT97.55.597.04.099.53.42,4⁃DNCB97.53.8101.53.596.52.92,6⁃DNCB96.04.195.03.498.02.7TNT101.55.3100.53.697.03.3
表5 不同分析方法用于测定水中硝基苯类物质的比较
Table 5 Comparison of different analytical methods for determination of nitroaromatics in aqueous samples
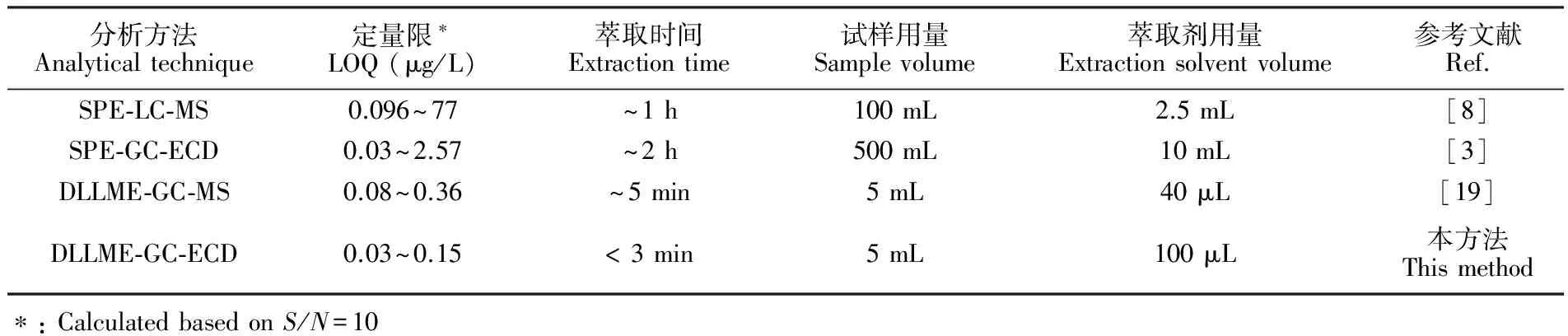
分析方法Analyticaltechnique定量限∗LOQ(μg/L)萃取时间Extractiontime试样用量Samplevolume萃取剂用量Extractionsolventvolume参考文献Ref.SPE⁃LC⁃MS0.096~77~1h100mL2.5mL[8]SPE⁃GC⁃ECD0.03~2.57~2h500mL10mL[3]DLLME⁃GC⁃MS0.08~0.36~5min5mL40μL[19]DLLME⁃GC⁃ECD0.03~0.15<3min5mL100μL本方法Thismethod∗:CalculatedbasedonS/N=10
1 Peng X T, Zhao X, Feng Y Q.J.Chromatogr.A,2011, 1218(52): 9314-9320
2 Zhang G J, Zhou X, Zang X H, Li Z, Wang C, Wang Z.ChineseChem.Lett.,2014, 25 (11): 1449-1454
3 ZOU Hai-Min, ZHOU Chen, YU Hui-Ju, ZHANG Qian, ZENG Hong-Yan, LI Yong-Xin.ChineseJ.Anal.Chem.,2016, 44(2): 297-304
邹海民, 周 琛, 余辉菊, 张 潜, 曾红燕, 李永新. 分析化学,2016, 44(2): 297-304
4 EPA 8091,NitroaromaticsandCyclicKetonesbyGasChromatography. US Environmental Protection Agency Standards
5 HJ 648-2013,WaterQuality:DeterminationofNitroaromaticsbyGasChromatography. National Environmental Protection Standards of the People's Republic of China
水质: 硝基苯类化合物的测定 液液萃取/固相萃取-气相色谱法. 中华人民共和国国家环境保护标准, HJ 648-2013
6 HJ 716-2014,WaterQuality:DeterminationofNitroaromaticsbyGasChromatographyMassSpectrometry. National Environmental Protection Standards of the People's Republic of China
水质: 硝基苯类化合物的测定 气相色谱-质谱法. 中华人民共和国国家环境保护标准, HJ 716-2014
7 Gaurav Kaur V, Kumar A, Kumar Malik A, Rai P K.J.Hazard.Mater.,2007, 147(3): 691-697
8 Rapp-Wright H, McEneff G, Murphy B, Gamble S, Morgan R, Beardah M, Barron L.J.Hazard.Mater.,2017, 329(1): 11-21
9 Reyes-Gallardo E M, Lasarte-Aragonés G, Lucena R, Cárdenas S, Valcárcel M.J.Chromatogr.A,2013, 1271(1): 50-55
10 Psillakis E, Kalogerakis N.J.Chromatogr.A,2001, 907(1): 211-219
11 Ebrahimzadeh H, Yamini Y, Kamarei F, Khalili-Zanjani M.Talanta,2007, 72(1): 193-198
12 Rezaee M, Assadi Y, Milani Hosseini M R, Aghaee E, Ahmadi F, Berijani S.J.Chromatogr.A,2006, 1116(1-2): 1-9
13 Berijani S, Assadi Y, Anbia M, Milani Hosseini M R, Aghaee E.J.Chromatogr.A,2006, 1123(1): 1-9
14 Yan H Y, Wang H.J.Chromatogr.A,2013, 1295(5): 1-15
15 Campillo N, Vias P,andrejovJ, Andruch V.Appl.Spectrosc.Rev.,2017, 52(4): 267-415
16 Ebrahimzadeh H, Yamini Y, Kamarei F.Talanta,2009, 79(5): 1472-1477
17 Sobhi H R, Kashtiaray A, Farahani H, Javaheri M, Ganjali M R.J.Hazard.Mater.,2010, 175(1): 279-283
18 Zhang D, Zeng X, Yu Z, Sheng G, Fu J.Anal.Methods,2011, 3(10): 2254-2260
19 Cortada C, Vidal L, Canals A.Talanta,2011, 85(5): 2546-2552
20 XIONG Jun, XIE Si-Long, LAI Yi-Dong.ChineseJournalofChromatography,2011, 29(2): 115-119
熊 珺, 谢思龙, 赖毅东. 色谱,2011, 29(2): 115-119
21 Jowkarderis M, Raofie F.Talanta,2012, 88(1): 50-53
22 Wu Y, Dai L, Cheng J, Guo F, Li J.Chromatographia,2010, 72(7-8): 695-699
23 Bahmaei M, Mashayekhi H A, Khalilian F.J.Braz.Chem.Soc.,2015, 26(7): 1475-1481
24 Ahmad U K, Mechor W H, Mohamed M.Anal.Lett.,2012, 45(15): 2198-2209
25 Fattahi N, Assadi Y, Hosseini M R M, Jahromi E Z.J.Chromatogr.A,2007, 1157(1): 23-29
26 Kozani R R, Assadi Y, Shemirani F, Hosseini M R M, Jamali M R.Talanta,2007, 72(2): 387-393
This work was supported by the National Natural Science Foundation of China (No. 21476177).
Determinationof15KindsofNitroaromaticsinAqueousSamples
UsingDispersiveLiquid-LiquidMicroextractionand
GasChromatographywithElectronCaptureDetection
DU Xiao-Di, LI Jun-Sheng, GUO Li-Ping, LEI Jia-Heng*
(DepartmentofChemistry,SchoolofChemistry,ChemicalEngineeringandLifeSciences,
WuhanUniversityofTechnology,Wuhan430070,China)
A method for the determination of 15 kinds of nitroaromatics in aqueous samples was developed by dispersive liquid-liquid microextraction and gas chromatography with electron capture detection. A high-density extractant applied in electron capture detector was screened out. The chromatographic conditions, types and dosages of extractants, types and dosages of dispersants, extraction time and the extraction temperature were optimized. The results showed that DB-35 capillary column had the best separation performance for the 15 kinds of nitroaromatics. The nitroaromatics could be separated within 22 min using programmed temperature control as follows: holding at an initial temperature of 80℃ and then heating to 180℃ at a ramping rate of 5℃/min. For the extraction of 15 kinds of nitroaromatics from 5 mL of aqueous sample, the extraction equilibrium could be reached within 30 s with a high extraction recovery of over 90% when using 100 μL of chlorobenzene as extracting solvent and 400 μL of methanol as disperser solvent. In addition, the enrichment factor could approach a high value of 45.0-48.8. The sediment collected by centrifugation was injected and analyzed by gas chromatography with electron capture detector. The limits of quantification of the developed method were 0.03-0.15 μg/L (S/N=10). The linear range was from 0.20 μg/L to 50.0 μg/L, while the correlation coefficients (R2) were more than 0.998. At the spiked level of 0.200 μg/L, the relative standard deviations of this method were 3.3%-8.9%, the relative recoveries ranged from 86.0% to 103.5%. At higher spiked level, the relative standard deviations were less than 5%, and the relative recoveries ranged from 94.5% to 101.5%.
Gas chromatography; Dispersive liquid-liquid microextraction; Nitroaromatics; Drinking water
9 August 2017; accepted 18 September 2017)
10.11895/j.issn.0253-3820.171160
猜你喜欢
环境卫生工程(2021年1期)2021-03-19 05:22:42
分析测试学报(2015年4期)2016-01-13 06:18:27
应用化工(2014年1期)2014-08-16 13:34:08
应用化工(2014年4期)2014-08-16 13:23:09
浙江科技学院学报(2014年6期)2014-02-28 22:12:09
中国氯碱(2014年10期)2014-02-28 01:04:59
河南科技(2014年8期)2014-02-27 14:07:42
环境工程技术学报(2012年6期)2012-08-22 14:10:39
中国烟草学报(2012年6期)2012-04-09 07:41:40
植物营养与肥料学报(2010年6期)2010-10-26 05:02:34
