
CiteSpace软件分析反刍动物胃肠道微生物研究科技论文的图谱
2017-10-23 06:59张国兴董刚辉李锡智王雅春
中国畜牧杂志 2017年10期
张国兴,董刚辉,李锡智,王雅春*
(1.中国农业大学动物科技学院,北京100193;2.北京首农畜牧发展有限公司,北京100029)
CiteSpace软件分析反刍动物胃肠道微生物研究科技论文的图谱
张国兴1,董刚辉2,李锡智2,王雅春1*
(1.中国农业大学动物科技学院,北京100193;2.北京首农畜牧发展有限公司,北京100029)
为了解反刍动物胃肠道微生物研究领域的研究历史,作者通过Web of Science数据库查找得到1940年到2017年2月20日为止6 372篇该领域的文献,通过CiteSpace软件分析其参考文献、关键词、作者、机构等元素,挑选重要的文献进行仔细阅读、分析、归纳和总结,展示该领域的研究方法发展史、研究内容发展史以及当前的研究热点。微生物研究方法在不断进步,人们对反刍动物胃肠道微生物的认识也越来越深入,研究者们不仅探究胃肠道微生物于宿主消化的意义,也开始发掘与宿主健康、生理、生产性状的直接联系。对于如何开展反刍动物胃肠道微生物的相关研究,建议全面收集相关研究信息、谨慎选择研究方法、有选择地研读科技文献。
反刍动物;胃肠道微生物;Cite Space
胃肠道微生物与人类的免疫、健康长寿、生理状态等密切相关,因此越来越受到人们的重视[1-2]。胃肠道微生物对反刍动物尤为重要,与单胃动物不同,反刍动物拥有庞大的前胃,其中瘤胃内微生物可降解纤维素,产生宿主能够利用的短链脂肪酸,可以利用非蛋白氮合成菌体蛋白供宿主利用,还能合成一些重要的维生素。瘤胃微生物总量约为反刍动物宿主细胞数量的100倍[3]。这些微生物与反刍动物的生产表现和健康息息相关[4]。反刍动物胃肠道微生物最早是动物营养相关领域研究的范畴,但是随着研究方法的进步和研究内容的深入,人们发现反刍动物胃肠道微生物的意义不仅在于饲料消化,还与许多生产性能相关,因此吸引了越来越多研究者的关注。
CiteSpace作为科学文献知识图谱分析软件,是在文献计量学基础上发展起来的一种文献分析技术,特色是绘制科学知识图谱,可视化显示知识资源及其关联。CiteSpace主要用4个指标来衡量文献的重要性,即Citation Counts、Bursts、Centrality、Sigma,分别解释为被引频次、引文突发性、中心度、中介中心性。Citation Counts即累计被引频次,它仅表示在领域内被引的频次,在CiteSpace的可视化图谱中其数值与节点圆大小呈正比;Bursts较高的文献,表示该文献在某一个确定时间段内被大量引用,在CiteSpace的可视化图谱中,这样的文献会用红色中心标示;Centrality和Sigma都衡量了这篇文献在该领域知识结构中的重要性,Centrality和Sigma较高的文献,是整个引文网络不可或缺的关联结点,在CiteSpace的可视化图谱中会用紫色外环标示[5]。Citation Counts、Bursts、Centrality、Sigma值 较 高的文献,在学科发展过程中作用较大,是学科的基础。
为了帮助同行了解反刍动物胃肠道微生物领域的研究概况,以便更加细致、全面地去设计试验,作者于2017 年2月20日在SCI 数据库中以“Topic: ("16S r*NA" or Microbi* or Metagenom*or Micro*Rganism)AND Topic: (Cow or Cattle or Beef or Dairy or Rumina*)AND Topic :(Rumen or Gastrointest* or Bowel or Gut or Gastric or Fec*or Intestin* or Enteric or Excrement or Faeces)”为检索式,共检索到6 372 条文献数据,其中,引文记录总计123 942 条;再借助CiteSpace来对这些科技文献进行分析,分析的基本数据单元包括参考文献、关键词、作者、机构,从而筛选重要文献进行阅读和总结。本文综述了该领域研究内容和方法的发展史、研究热点、研究主体,并提供一些研究建议。
1 研究历史分析
1.1 反刍动物胃肠道微生物研究的国际发文量分析如图1所示,该领域在1954年发表了第1篇文献,1954—1990年,平均每年发文量约2篇(图1中未显示)。1990—2005年,发文量处于缓慢增长时期;2005年以后,发文量快速增长;2016年发文量已达556篇。
1.2 对反刍动物胃肠道微生物区系结构的探索 图2是CiteSpace生成的胃肠道微生物领域高被引文献按照时间排序的共被引网络知识图谱,每一个节点都是在本领域被引用次数较高的文献;连线代表共被引关系,线越粗,共被引次数越高。图2中,共有243篇文献和485条连线,标出名字的文献是被引次数最高的4篇。被引频次最高的5篇文献的详细信息如表1所示。被引频次较高的文献对该领域研究进展贡献较大,是该领域的学科基础,主要探讨了领域内重要的分析方法,也探索了反刍动物胃肠道重要的微生物群体,进而完善反刍动物胃肠道微生物的组成结构信息。
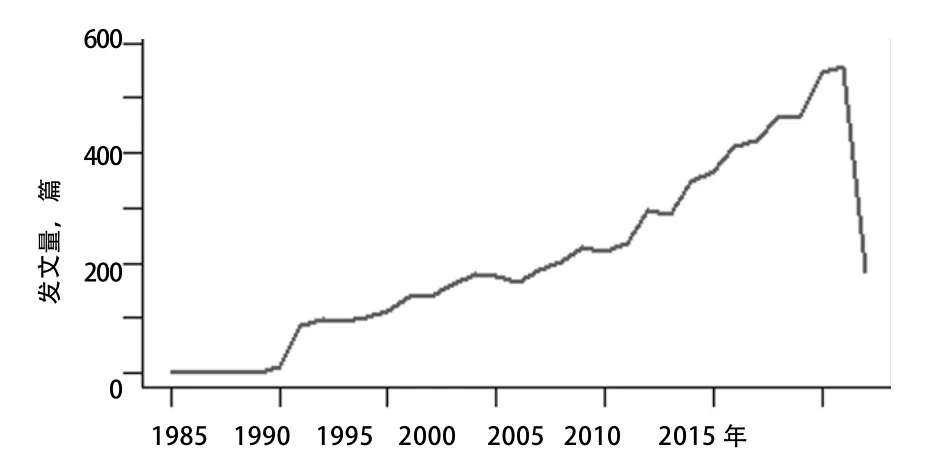
图1 反刍动物胃肠道微生物研究国际年发文量
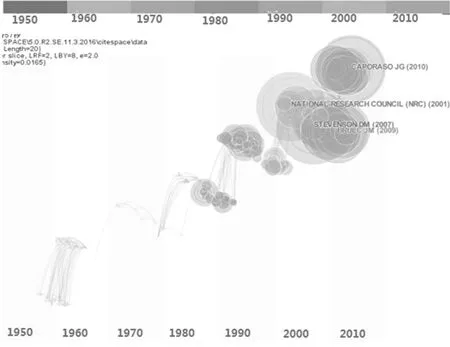
图2 反刍动物胃肠道微生物领域文献共被引网络知识图谱
在体外分离培养和发酵活动研究的时代,完整地阐释反刍动物胃肠道微生物区系结构几乎是不可能的,只能分离、培养和研究可培养的微生物,如瘤胃球菌属、甲烷细菌属和丁酸弧菌属等[6]。甲烷不能被反刍动物利用,如瘤胃中产甲烷菌较多,将会造成饲料利用效率下降;如果甲烷排放到大气中,会加重温室效应;产甲烷菌成为后续研究的焦点。后肠是发酵产生甲烷的重要场所,Stewart[7]对后肠微生物的阐释具有重要意义;Janssen等[8]提出古细菌主要是由产甲烷菌组成的(被引频次104);Hook等[9]总结了古细菌和后肠道微生物是主要的产甲烷微生物。Stevenson等[10]认为瘤胃微生物主要是由普氏菌属(42%~60%)和其他稀有菌属(少于1%)构成(被引频次135),这种瘤胃微生物区系结构的阐释是不够完全的。近年发现,瘤胃微生物主要由细菌、真菌、原虫、古细菌组成[11]。Brulc等[12]发现细菌占到所有微生物的95%。目前高通量测序技术用于胃肠道微生物区系结构的阐释进展很快,Jami等[13]研究肉牛瘤胃微生物区系结构发现,瘤胃微生物细菌界中厚壁菌门、拟杆菌门、变形菌门、放线菌门和软壁菌门分别占42%、51%、5.21%、0.87%和0.68%,而其他细菌非常少,且个体间瘤胃细菌区系组成结构只有51%的相似度。DeOliveira等[14]用高通量测序技术研究了巴西内罗尔(Nelore)阉牛整个胃肠道(前胃、小肠及大肠)的微生物区系,发现胃肠道不同区段的微生物区系结构存在很大差异,但是厚壁菌门、拟杆菌门、浮霉菌门和螺旋体属是3个区段共有的细菌。当下,胃肠道微生物区系结构的个体差异及影响因素引起研究者关注,区系结构的探索也更加细致。有的研究对瘤胃液进行过滤,只研究瘤胃液的液相或者固相中的微生物;有的将液相和固相中微生物混在一起研究。研究表明,瘤胃液固相中的微生物群是瘤胃液中一个重要的、特殊的微生物群体,尤其对研究饲料的微生物消化过程具有重要意义,例如Brulc等[12]对固体纤维上的微生物进行了研究,发现饲料进入瘤胃后,最先在饲料上定植的是能够降解支链多糖的微生物,这些微生物很难去降解纤维素(被引频次109)。

表1 反刍动物胃肠道微生物领域被引频次排前5的文献
1.3 研究方法的纵向分析 反刍动物胃肠道微生物研究离不开一些基本的营养学分析方法以及原则,如表1中提到的NRC奶牛营养需要[15],以及Van Soest纤维分析方案[16],AOAC推荐的化学成分分析方法[17]。
反刍动物微生物分析方法的发展大致可以分为3个时代,即体外分离培养和发酵活动研究时代、分子生物学技术时代、二代高通量测序时代。表1中文章标题里提到的PCR和QIIME实际上就是分子生物学时代和二代高通量测序时代的缩影。
在20世纪90年代以前,人们对反刍动物胃肠道微生物的认识,主要依靠对少数几种可培养微生物进行体外分离培养,发酵活动研究以及分类命名[18]。瓶颈在于很难涉及稀有的、不可培养的微生物;瘤胃微生物大都是厌氧型,即使能够培养,其对培养条件的要求也非常苛刻[19];当时,微生物的分类主要依靠对微生物形态的观察,但是形态特征的进化和系统发育学意义很难去阐释[18];再者,肉眼计数难度很大,且不准确。
早在1965年,Zuckerkandl等[20]就预测了大分子序列在微生物分类学方面的运用;1980年前后,人们开始尝试运用各种大分子的序列变异信息研究微生物分类,其中较有代表性的包括质粒DNA、染色体DNA、核糖体RNA(包括大亚基23S和5S,小亚基16S)等[18]。 Woese[21]注意到16S rRNA存在可变区和保守区,初步阐释了16S rRNA可变区信息更具微生物分类意义。1990年前后发展出许多微生物分子生物学研究技术,如PCR、荧光原位杂交技术、凝胶电泳、限制片段长度多态性、印记杂交技术等,大多基于16S rRNA微生物分类学,White等[22]总结了其中几种在反刍动物微生物研究中的应用。定量PCR技术常基于16S rRNA微生物分类学用于检测样品中特定微生物,通过菌种特异性的引物,定量PCR可以对已知菌种进行定量分析。但是研究胃肠道微生物的区系结构光靠PCR技术、通量和速度还不够,且不易研究未知的微生物物种。
Caporaso等[23]提出用QIIME软件分析16S rDNA(16S rRNA基因)高通量测序结果,进而分析微生物多样性(被引频次123)。自此之后,涌现了大量关于反刍动物胃肠道微生物区系结构的研究。16S rDNA高通量测序分析微生物的优点是既能对已知或者未知的微生物进行高通量定性和定量分析,也能进行粗略的功能分析;二代高通量技术不仅能够测定16S rDNA序列,也能够对微生物组的所有DNA进行测序(宏基因组测序)。由于宏基因组不仅包括分类学信息,还包含了基因信息,因此不仅能够对微生物进行定性和定量分析,还能进行深入的功能探究;二代高通量测序技术在胃肠道微生物研究领域的运用,也使得微生物组(Micobiome)、微生物区系(Micobiota)、多样性(Diversity)、宏基因组(Metagenome)等概念大量被研究者们运用。当然,高通量技术也有其局限性,由于通量较大,分析菌种过多,如果要专门检测某个菌种,其特异性和灵敏度不如定量PCR[24],且成本较高。
1.4 研究内容的纵向分析 CiteSpace提供了文献聚类功能,可以根据文献间的内容关联程度对一些重要的文献进行聚类,并计算类别内最能代表这些文献关注点的短语,作为这个类别的名称,同时也能够导出每个聚类内的具体文献成员,聚类大小反映了聚类内文献成员的数量。表2列出了所有聚类大小大于5的文献聚类。
1956—1963年主要研究瘤胃微生物对饲料的作用,重点是微生物。在体外研究微生物的发酵活动是当时的热点,如聚类“Microbial Digestion”中,Bryant[6]总结了当时的研究方法和主要研究成果,对能够体外培养的瘤胃微生物进行培养条件、发酵底物、发酵产物、发酵参数的研究。
1965—1973年是本领域发展的瓶颈时期,而1973—1990年主要研究饲料对微生物发酵的影响,进而研究饲料的利用效率,重点是饲料。这段时间的重要文献聚类“Steam Cooking,Sucrose Supplement,Various Nitrogen Supplementation”等分析了饲料的处理方式以及能量、蛋白的来源影响了微生物发酵利用的效率,开始关注可消化蛋白质、非结构碳水化合物、可溶淀粉、氨基酸、非蛋白氮等六大主要营养物质的细化分类的微生物利用过程,如 聚 类“Various Nitrogen Supplementation” 中,Stokes等[25]发现非结构碳水化合物和可降解蛋白的比例影响了瘤胃微生物对饲料的利用效率。1990年出现的一个比较大的聚类“Comparative Feeding Value”充分拓展这一研究内容,不同来源营养物质的微生物利用效率不同,出现相对饲用价值这一概念。
1990—2010年,随着研究手段的更新,该领域涌现了许多比较大的研究聚类。1997年1个名为“Targeted Oligonucleotide Probe”的较大聚类探讨了这个时期检测微生物的分子生物学手段之一——寡核苷酸探针。该时期对一些重要营养物质的代谢和微生物、饲料利用率的关系作了更加细致的研究;其中,聚类“Purine Derivative”中文献围绕嘌呤衍生物这一微生物蛋白合成的标记代谢物展开了一系列研究;聚类“Lipid Metabolism”中的文章探讨了脂类代谢和胃肠道微生物的关系;聚类“Rumen Methanogen”中的文献探讨了甲烷代谢、胃肠道微生物、饲料利用效率、温室效应之间的关系;聚类“Amino Acid”探讨的核心问题是如何降低氨气的释放速率和增加微生物蛋白的合成[26],与甲烷类似,氨气释放过快,会导致蛋白质饲料的浪费以及环境污染。聚类“Essential Oil”中的文献主要探讨了一些植物提取物(含精油及其代谢物)的抗菌促生长作用,如Chaves[27]探讨了精油成员之一——香荆芥酚对幼龄羊生长过程中采食量、瘤胃微生物发酵、生长性能、胴体品质的影响。
2010年以后,由于高通量测序技术应用于微生物研究领域,微生物相关研究不再局限于动物营养相关范畴。微生物区系结构非常复杂且受很多因素影响,但是在同一个物种内,饲料的影响仍然是最主要的。2010年的聚类“Gut Microbiome”中的文献分析了微生物组和反刍动物饲料[28]、遗传[29]、围产[30]、年龄[31]、健康[32]等因素的关系。
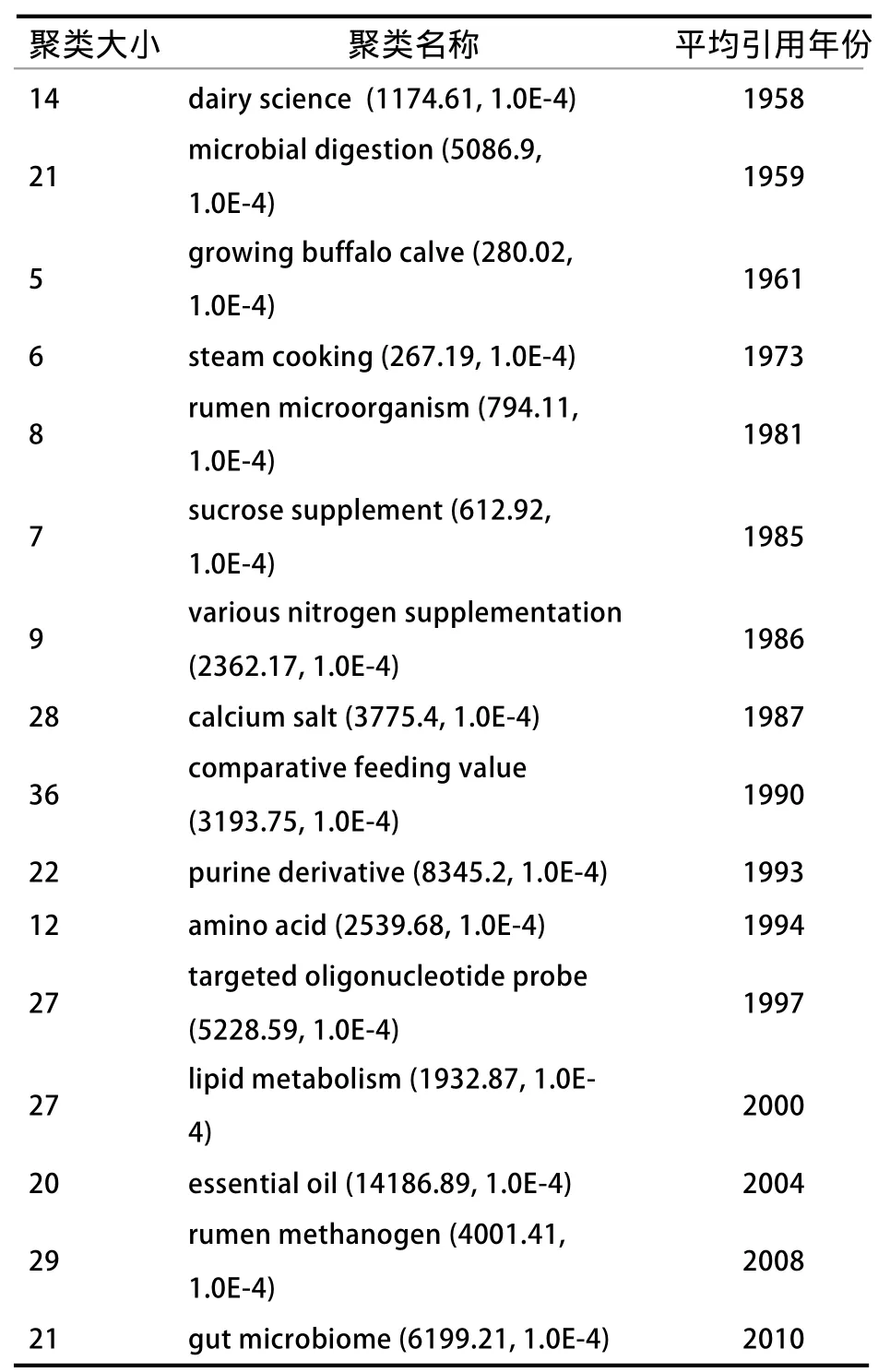
表2 包含文献数量最多的16个文献聚类
图3显示了本领域文献中引用突发值最高的20个关键词。从关键词爆发年份区间来看,研究内容的发展历史基本与前述一致。1940—1990年,出现较多的关键词是Invitro(体外)、Nitrogen(氮)、Amino-acid,其中,由关键词“体外”可以窥见这段时间体外分离培养、发酵的研究手段;1990—2000年,出现较多的关键词是Passage(通过瘤胃的营养物质量)、Protein、Nitrogen、Small-intestine(小肠),可窥见这段时间重点是研究营养物质的代谢及其被反刍动物利用的效率;2010年以后,大量热词出现了,诸如Diversity、Gut Microbiota、Microbime等关键词彰显了高通量测序技术带来的变革,Methane、Emissions(甲烷或氨气的排放)、Methane Production(甲烷产量)也表明这个时期的研究重点是胃肠道微生物和甲烷、氨气排放以及饲料利用效率、环境之间的关系。
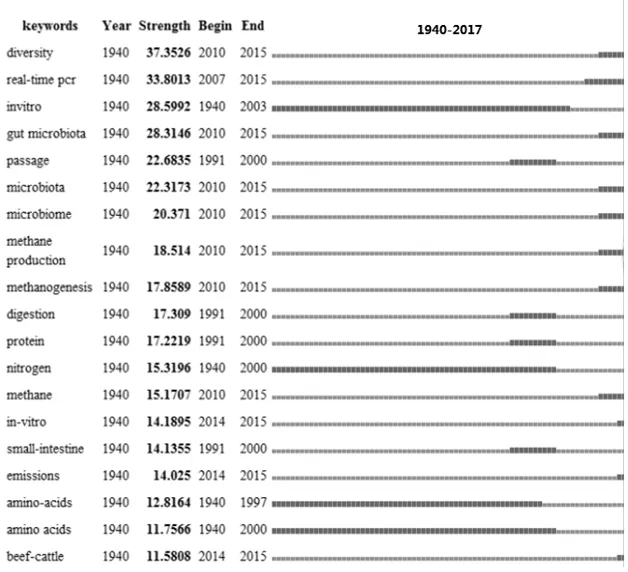
图3 引用突发值最高的20个关键词
2 研究主体分析
2.1 发文量较高的作者 图4中每个节点代表1个作者,节点大小代表该作者发文量多少,节点间的连线代表作者间的合作关系(在同一篇文献中出现)。图4包括112个作者和214次合作关系,说明该领域内研究者间相互引用、相互合作较频繁。图中标出名字的是在该领域内发文数量最多的6位作者,对本领域的贡献较大。其中McAllister TA发文量最高(110篇),McAllister TA和Newbold CJ的Sigma值较高,这些作者对这个领域的研究有重要贡献。
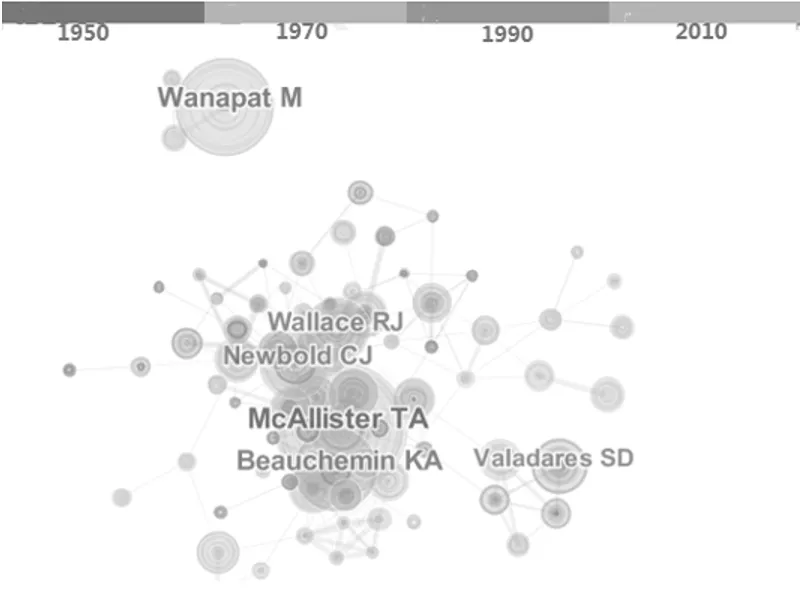
图4 发文量较高的作者合作网络图谱
2.2 发文量较高的机构 本文查阅的文献共涉及84个机构和209条机构间连线,说明该领域内各机构的合作较多。由表3可知,该领域发文量最高的9个机构分别来自加拿大(2)、美国(4)、法国(1)、巴西(1)和泰国(1)。其中美国俄亥俄州立大学、加州大学戴维斯分校和加拿大农业及食品部等机构中心度较高,其研究成果在本领域较有影响力。
3 关于反刍动物胃肠道微生物研究的几点建议
3.1 全面收集研究相关信息 胃肠道微生物组与宿主的共生互作关系极其复杂。研究表明,肉牛个体间瘤胃细菌区系组成结构的相似度只有51%[13],而变异由很多因素造成。宿主会作用于胃肠道微生物组,奶牛的年龄、遗传背景、泌乳阶段、围产、饲料、饲料添加剂、益生素、抗生素、疾病、瘤胃pH等因素都会影响反刍动物的胃肠道微生物区系结构[33-36],而胃肠道微生物的区系结构又会反过来作用于反刍动物的生产性状(饲料利用效率、产奶量、乳蛋白、乳脂等)以及次级性状(疾病)[4,37-38],外界环境也会影响反刍动物的胃肠道微生物,如地理[39]、环境温度[40]。在其他物种的研究中(尤其是人类),发现胃肠道微生物组和宿主的寿命、免疫系统的建立和维持、肥胖、抑郁、早期成长环境、家庭生活习惯、地域等因素有关[1,41-44]。虽然宿主与胃肠道微生物组的互作以及因果关系尚不明朗,但足以启示研究者在进行反刍动物胃肠道微生物的相关研究时,必须全面收集信息,例如奶牛营养条件、年龄、胎次、泌乳阶段、体况、遗传背景、生理状态(怀孕、体温、瘤胃pH、生理生化指标)、疾病记录、用药记录和生产表现等,对可能的影响因素进行控制或者设置对照。

表3 反刍动物胃肠道微生物领域发文较多的9个机构
3.2 方法选择需谨慎 研究反刍动物胃肠道微生物的方法主要包括体外分离培养与发酵、分子生物学手段、高通量测序手段3大类,每种方法都有自己的特点和优势,要根据试验设计进行比较选择。如果选择高通量测序手段来研究反刍动物胃肠道微生物,就会涉及采样方法、样品保存方法、DNA的提取方法、测序平台、16S rDNA测序高变区段、测序量、分析软件等细节方法的选择,这些细节方法的选择将会影响最终的分析结果,例如:①瘤胃液的采集方法主要有瘤胃液采集管和瘤胃瘘管2种,瘤胃瘘管手术困难但是方便长期对同一头牛取瘤胃液,瘤胃液采集管则便于大群体一次性采集瘤胃液;②DNA提取方法较多,Henderson等[45]通过对不同采样方法、DNA提取方法进行对比,发现不同采样方法、DNA提取方法之间的分析效果有细微区别,应根据实验设计、测序量、测序方法选择采样方法和DNA提取方法;③样本保存一般选择液氮罐或者-80℃冰箱,也可使用DNA样品保护剂来降低保存难度;④目前常用测序平台包括hiseq和miseq,miseq的优点是测序长度长、仪器成本低,但是测序通量小,hiseq的优点是测序通量大,但是测序长度偏低、仪器成本高,目前hiseq的PE 250策略的测序长度已经能够和miseq媲美;⑤16S rDNA全长1 540 bp,有9个可变区,不同物种、不同可变区的组合效果存在差别[46-47],奶牛的研究多选用V3+V4区测序;⑥关于16S rDNA测序量,Lundin等[48]研究454 测序数据,认为每个样品1 000 条序列可反映90% 的β多样性信息,而每个样品5 000 条序列才能反映90%的α多样性信息,当然,不同样本类型对测序量的要求也不一样。具体研究方案、测序方案和生物信息分析策略的选择,也可查阅李东萍等[49]的综述。
3.3 有选择地深入研读他人研究成果 由于反刍动物胃肠道微生物领域目前比较热门,几乎每天都有新的研究成果发表,但是很多研究尚存在局限性。一是历史的局限性:领域内的研究方法正在快速更新换代,知识结构也在变化,故研读论文要结合历史背景,不可太片面;二是试验设计存在缺陷:由于样本量较少,考虑因素不全面,研究成果需要进一步验证。本文中提供了一些较权威的作者、机构和论文,供同行查阅。
[1] Macpherson A J, Harris N L. Interactions between commensal intestinal bacteria and the immune system[J].Nat Rev Immunol, 2004, 4:478-485.
[2] Biagi E, Franceschi C, Rampelli S, et al. Gut microbiota and extreme longevity[J]. Curr Biol, 2016, 26: 1480.
[3] Attwood G T, Kelly W J, Altermann E H, et al. Application of rumen microbial genome information to livestock systems in the postgenomic era[J]. Aust J Exp Agr, 2008,48(6-7): 695-700.
[4] Malmuthuge N, Guan L L. Understanding host-microbial interactions in rumen: searching the best opportunity for microbiota manipulation[J]. J Anim Sci Biotechnol, 2017,8:8.
[5] 曹露, 王泽昭, 董刚辉, 等. 奶牛温度应激研究知识图谱分析[J]. 畜牧兽医学报, 2015, (9): 1489-1495.
[6] Bryant M P. Symposium on microbial digestion in ruminants identification of groups of anaerobic bacteria active in rumen[J]. J Anim Sci, 1963, 22(3): 801.
[7] Stewart C S. Microorganisms in hindgut fermentors[M].England: Chapman and Hall, 1997, 142-186.
[8] Janssen P H, Kirs M. Structure of the archaeal community of the rumen[J]. Appl Environ Microb, 2008, 74(12): 3619-3625.
[9] Hook S E, Wright A D, Mcbride B W. Methanogens:methane producers of the rumen and mitigation strategies[J]. Archaea, 2010: 945785.
[10] Stevenson D M, Weimer P J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR[J]. Appl Microbiol Biot, 2007, 75(1): 165-174.
[11] Flint H J. The rumen microbial ecosystem--some recent developments[J]. Trends Microbiol, 1997, 5(12): 483-488.
[12] Brulc J M, Antonopoulos D A, Miller M E B, et al. Genecentric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases[J].P Natl Acad Sci USA, 2009, 106(6): 1948-1953.
[13] Jami E, Mizrahi I. Composition and similarity of bovine rumen microbiota across individual animals[J]. PLoS One,2012, 7(3): e33306.
[14] De Oliveira M N, Jewell K A, Freitas F S, et al.Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer[J]. Vet Microbiol, 2013,164(3-4): 307-314.
[15] National Research Council Subcommittee on Dairy Cattle Nutrition. Nutrient requirements of dairy cattle[M]. USA:National Academy Press, 2001, 381.
[16] Van Soest P J, Robertson J B, Lewis B A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition[J]. J Dairy Sci, 1991, 74(10): 3583-3597.
[17] Helrich K C. Official Methods of Analysis of the AOAC[M].USA: Association of Official Analytical Chemists, 1990,684.
[18] Krause D O, Russell J B. How many ruminal bacteria are there?[J]. J Dairy Sci, 1996, 79(8): 1467-1475.
[19] Hungate R E, Smith W, Clarke R T. Suitability of butyl rubber stoppers for closing anaerobic roll culture tubes[J].J Bacteriol, 1966, 91(2): 908.
[20] Zuckerkandl E, Pauling L. Molecules as documents of evolutionary history[J]. J Theor Biol, 1965, 8(2): 357.
[21] Woese C R. Bacterial evolution[J]. Microbiol Rev, 1987,51(2):221-271.
[22] White B A, Cann I, Kocherginskaya S A, et al. Molecular analysis of archaea, bacteria and eucarya communities in the rumen - Review[J]. Asian-Austral J Anim, 1999, 12(1):129-138.
[23] Caporaso J G, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nat Methods, 2010, 7(5): 335-336.
[24] Wang F, Huang G, Cai D, et al. Qualitative and semiquantitative analysis of fecal Bifidobacterium species in centenarians living in Bama, Guangxi, China[J]. Curr Microbiol, 2015, 71(1): 143-149.
[25] Stokes S R, Hoover W H, Miller T K, et al. Ruminal digestion and microbial utilization of diets varying in type of carbohydrate and protein[J]. J Dairy Sci, 1991, 74(3):871.
[26] Elizalde J C, Merchen N R, Faulkner D B. Supplemental cracked corn for steers fed fresh alfalfa: II. Protein and amino acid digestion[J]. J Anim Sci, 1999, 77(2): 467-475.[27] Chaves A V, Stanford K, Gibson L L, et al. Effects of carvacrol and cinnamaldehyde on intake, rumen fermentation, growth performance, and carcass characteristics of growing lambs[J]. Anim Feed Sci Technol, 2008, 145(1-4): 396-408.
[28] Kong Y, Teather R, Forster R. Composition, spatial distribution, and diversity of the bacterial communities in the rumen of cows fed different forages[J]. FEMS Microbiol Ecol, 2010, 74(3): 612-622.
[29] Hernandez-Sanabria E, Goonewardene L A, Wang Z, et al. Influence of sire breed on the interplay among rumen microbial populations inhabiting the rumen liquid of the progeny in beef cattle[J]. PLoS One, 2013, 8(3): e58461.
[30] Pitta D W, Kumar S, Vecchiarelli B, et al. Temporal dynamics in the ruminal microbiome of dairy cows during the transition period[J]. J Anim Sci, 2014, 92(9): 4014-4022.
[31] Klein-Joebstl D, Schornsteiner E, Mann E, et al.Pyrosequencing reveals diverse fecal microbiota in Simmental calves during early development[J]. Front Microbiol, 2014, 5(5):622.
[32] Ross E M, Moate P J, Marett L C, et al. Metagenomic predictions: from microbiome to complex health and environmental phenotypes in humans and cattle[J]. PLoS One, 2013, 8(9): e73056.
[33] Patel V, Patel A K, Parmar N R, et al. Characterization of the rumen microbiome of Indian Kankrej cattle (Bos indicus) adapted to different forage diet[J]. Appl Microbiol Biotechnol, 2014, 98(23): 9749-9761.
[34] Pitta D W, Kumar S, Vecchiarelli B, et al. Temporal dynamics in the ruminal microbiome of dairy cows during the transition period[J]. J Anim Sci, 2014, 92(9): 4014-4022.
[35] Pinloche E, Mcewan N, Marden J, et al. The effects of a probiotic yeast on the bacterial diversity and population structure in the rumen of cattle[J]. PLoS One, 2013, 8:e678247.
[36] Jewell K A, Mccormick C A, Odt C L, et al. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency[J]. Appl Environ Microb, 2015, 81(14): 4697-4710.
[37] Zhang J, Xu C, Huo D, et al. Comparative study of the gut microbiome potentially related to milk protein in Murrah buffaloes (Bubalus bubalis) and Chinese Holstein cattle[J].Scientific Reports, 2017, 7: 42189.
[38] Bath C, Morrison M, Ross E M, et al. The symbiotic rumen microbiome and cattle performance: a brief review[J].Anim Product Sci, 2013, 53(9): 876-881.
[39] Rira M, Morgavi D P, Popova M, et al. Ruminal methanogens and bacteria populations in sheep are modified by a tropical environment[J]. Anim Feed Sci Technol, 2016, 220: 226-236.
[40] Wang J, Wang J, Bu D. Quantities of ruminal cellulytic bacteria in heat stress dairy cattle[J]. Journal of China Agricultural University, 2011, 16(4):102-106.
[41] Koenig J E, Spor A, Scalfone N, et al. Succession of microbial consortia in the developing infant gut microbiome[J]. P Natl Acad Sci USA, 2011, 1081:4578-4585.
[42] Hehemann J, Correc G, Barbeyron T, et al. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota[J]. Nature, 2010, 464(7290):123-908.
[43] Maynard C L, Elson C O, Hatton R D, et al. Reciprocal interactions of the intestinal microbiota and immune system[J]. Nature, 2012, 489(7415): 231-241.
[44] Smith P, Willemsen D, Popkes M L, et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish[J]. ELIFE, 2017, 6: e27014.
[45] Henderson G, Cox F, Kittelmann S, et al. Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities[J]. PLoS One, 2013, 8: e747879.
[46] Claesson M J, Wang Q, O'Sullivan O. Comparison of two next-generatio n sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions[J]. Nucleic Acids Res,2010, 22(38): e200.
[47] Kozich J J, Westcott S L, Baxter N T, et al. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the Miseq Illumina sequencing platform[J]. Appl Environ Microb, 2013,79(17): 5112-5120.
[48] Lundin D, Severin I, Logue J B, et al. Which sequencing depth is sufficient to describe patterns in bacterial alphaand beta-diversity[J]. Env Microbiol Rep, 2012, 4(3):367-372.
[49] 李东萍, 郭明璋, 许文涛. 16S rRNA测序技术在肠道微生物中的应用研究进展[J]. 生物技术通报, 2015, (2):71-77.
An Atlas Analysis of the Scientific Literatures on Ruminal Gastrointestinal Microbes Using CiteSpace
ZHANG Guo-xing1, DONG Gang-hui2, LI Xi-zhi2, WANG Ya-chun1*
(1. College of Animal Science and Technology, China Agricultural University, Beijing 100193, China;2. Beijing Sunlon Animal Husbandry Development Co.LTD, Beijing 100029, China)
To understand the developmental history of the study based on ruminal, gastrointestinal microbes, by using Web of Science, authors found 6372 scientific literature of the year ranges from 1940 to 20 February 2017. The references, key words, authors and institutions of the scientific literature were analyzed by using CiteSpace software which enable the authors to select the most influential and important scientific literature to read, analyzes, and summarize carefully. In this review, the authors characterized the developmental history of research methods, contents about ruminal gastrointestinal microbes and introduced some popular topics in this field. Research techniques of microbes keep advancing, people learn more and more about ruminal gastrointestinal microbes. Researchers study not only the significance of gastrointestinal microbes to host digestion, but also the direct relationship between gastrointestinal microbes and host health, physiology,and production traits. In order to characterize the ruminal gastrointestinal microbes convectively, authors suggested a study to collect the information about the objective of research thoroughly, choose the techniques to use in research study carefully, and read the scientific literature selectively.
Ruminant; Gastrointestinal microbes; CiteSpace
S814
A
10.19556/j.0258-7033.2017-10-004
2017-05-24;
2017-08-01
现代农业(奶牛)产业技术体系建设专项资金(CARS-36);长江学者和创新团队发展计划(IRT_15R62)作者简介:张国兴(1994-),男,云南昆明人,硕士研究生,主要从事奶牛胃肠道微生物研究,E-mail: 947366452@qq.com
*通讯作者:王雅春,博士,教授,主要从事动物分子数量遗传学研究,E-mail: wangyachun@cau.edu.cn
猜你喜欢
吉林畜牧兽医(2022年6期)2022-11-16
热带作物学报(2022年9期)2022-10-17
中国典型病例大全(2022年11期)2022-05-13
云南农业科技(2021年6期)2021-12-30
养殖与饲料(2021年6期)2021-11-30
中国动物保健(2020年6期)2020-11-27
透析与人工器官(2020年1期)2020-11-16
科学(2020年2期)2020-08-24
铁道通信信号(2019年8期)2019-10-10
中国现代中药(2019年5期)2019-07-03
