
弱磁场强化零价铁对水中U(Ⅵ)去除效能
2017-10-14 03:55曹贝李锦祥关小红
化工学报 2017年8期
曹贝,李锦祥,关小红
弱磁场强化零价铁对水中U(Ⅵ)去除效能
曹贝,李锦祥,关小红
(同济大学环境科学与工程学院,上海 200092)
考察了弱磁场(WMF)对零价铁去除水中U(Ⅵ)效能的影响,并探讨了其主要机理。结果表明,在初始pH(pHini)为3.0~7.0,有弱磁场(w/ WMF)的条件下,零价铁去除U(Ⅵ)的一级动力学速率常数提高了0.7~11.2倍。当初始pH为4.0、零价铁的投加量为0.5 g·L-1时,弱磁场作用下的零价铁对U(Ⅵ)的去除容量为1.7 g·g-1,相比无弱磁场(w/o WMF)时提高了约0.3倍。pH变化、Fe2+浓度和SEM的结果说明,弱磁场通过促进零价铁的腐蚀促进其对U(Ⅵ)的去除。从XPS光谱分析中发现,零价铁去除U(Ⅵ)的主要机理为先吸附、后还原。弱磁场只能够加速其吸附和还原过程,而不能影响零价铁对U(Ⅵ)的去除机理。弱磁场促进零价铁去除U(Ⅵ)具有价格低廉、环境友好、无须额外的能量和药剂投入等优点,因而有良好的应用前景。
弱磁场;零价铁;U(Ⅵ);动力学;腐蚀;吸附;去除容量
引 言
随着核工业的发展,人们对铀的开发和利用越来越关注。有研究表明,到2020年,仅核电工业对天然铀的消耗量可达到3.34万吨,到2050年,电工业对天然铀的消耗量将达到407万吨以上[1]。然而,铀在开发和利用过程中,会对环境造成污染和破坏。根据调查结果,在中国柴达木盆地尕斯库勒湖、大柴旦湖和希里沟湖等盐湖卤水中铀的含量是海水中铀含量的几十倍甚至几百倍,有些盐湖晶间卤水中的铀含量高达40 mg·L-1[2]。Placzek等[3]对得克萨斯州南海线的地下水进行调查,发现部分地下水的铀含量高达10 mg·L-1。由于铀是重金属,所以它的化学毒性要比辐射危害大很多[4]。长期暴露在含铀的环境中,容易造成人的肝、肾等功能损伤,甚至会造成死亡[5]。
水中的铀有3种价态:U(0)、U(Ⅳ)和U(Ⅵ)。U(0)和U(Ⅳ)不易迁移,对水环境影响较小。U(Ⅵ)可以与水中多种离子配位,迁移性较大,易对环境造成污染[2]。近年来,很多学者对U(Ⅵ)的去除方法进行了研究,如吸附法、生物法、吸附还原法等,然而这些方法存在着去除效能不佳、操作复杂、成本高、潜在的环境危害等缺陷。零价铁由于其价格低廉、来源广泛、环境友好、易于操作等优势,已经在地下水修复、工业废水处理等方面展现出巨大潜力[6]。已有学者应用零价铁实现了对U(Ⅵ)的快速去除[7-8],其研究重心集中在对U(Ⅵ)的去除速率上,关于零价铁对U(Ⅵ)的去除容量研究较少,而去除容量在实际应用中和零价铁的投加量密切相关,具有一定的研究价值。
另一方面,由于制备过程中经历高温,零价铁表面都会覆盖一层铁氧化物膜,导致一般的微米零价铁反应活性比较低。很多学者关于如何提高零 价铁的反应活性做了大量的研究,如酸洗预处 理[9]、氢气预处理[10]、超声预处理[11]、外加金属离子[12-13]、合成纳米零价铁[14]、合成铁基双金属材 料[15]等。尽管这些方法能够在一定程度上提高零价铁的活性,但是其在实际应用上仍存在着成本高、操作复杂、潜在的生态毒性等方面的缺陷[6]。有研究表明弱磁场作为一种环保、无毒、无须额外的能源和药剂投入的方法,能够强化零价铁对溶液中Se(Ⅳ)[16]、As(Ⅲ,Ⅴ)[17]、Cr(Ⅵ)[18]、Cu(Ⅱ)[19]、Sb(Ⅴ)[20]等有毒(类)金属离子的去除,而关于 弱磁场能否促进零价铁对U(Ⅵ)的去除仍然有待探究。
基于以上讨论,本研究在去除速率和去除容量两方面,考察弱磁场对零价铁去除U(Ⅵ)产生的影响,并通过与其他去除U(Ⅵ)技术的对比,为含有U(Ⅵ)水的处理提供指导。
1 实验材料与方法
1.1 材料与试剂
实验中所用的还原性铁粉(98%)购自上海市金山冶炼厂。U(Ⅵ)[UO2(NO3)2·6H2O(≥99%)]购自湖北楚盛威化工有限公司,并用超纯水配制浓度为20 mmol·L-1U(Ⅵ)储备液。搅拌设备为机械搅拌器(400 r·min-1)。用小磁片在广口瓶底部进行加磁。实验所用的玻璃仪器均用硝酸浸泡10 h,再用蒸馏水及超纯水分别清洗3次,烘干后备用。
1.2 实验过程
本实验在500 ml的广口瓶中进行,敞口,水浴控温为25℃。在反应初始阶段用NaOH和HCl调节至所需pH,整个实验过程中无缓冲,也不再调节pH。实验结束后,通过抽滤设备,取得反应后固体,真空冷冻干燥后,无氧密封,供后续的SEM和XPS测试使用。
零价铁去除U(Ⅵ)动力学实验采用的反应初始pH为3.0、4.0、5.0、6.0、7.0;在初始pH为3.0和4.0时,反应时间控制在2 h;初始pH为5.0、6.0、7.0时,反应时间控制在3 h。零价铁的投加量为1.0 g·L-1,U(Ⅵ)的初始浓度为200 μmol·L-1,背景离子为0.01 mol·L-1NaCl。
初始U(Ⅵ)浓度为4 mmol·L-1的实验中,零价铁的投加量为0.5 g·L-1,背景离子为0.01 mol·L-1NaCl,初始pH用NaOH和HCl调节至4.0;反应时间为38 h,在30 h时实验达到平衡。以此为基础,只改变U(Ⅵ)的初始浓度,反应进行30 h达到平衡后计算零价铁去除U(Ⅵ)的容量。
1.3 测试方法
U(Ⅵ)和Fe2+离子浓度用双光束紫外可见分光光度计(TU-1901)进行测量。U(Ⅵ)显色剂采用偶氮胂Ⅲ,测定波长在652 nm处。二价铁的显色剂采用邻菲啰啉,测定波长在510 nm处。用Phenom台式扫描电子显微镜(SEM)观察固体产物的表面形貌。为了考察零价铁的腐蚀产物和表面铀元素的存在价态,采用X射线光电子能谱(PHI 5000 VersaProbe Ⅱ)对固体产物表面进行分析,并用XPS peak fit软件进行拟合。
2 实验结果与讨论
2.1 pH对零价铁去除U(Ⅵ)的影响
当初始pH为3.0~7.0时,外置弱磁场均能提高零价铁对U(Ⅵ)的去除速率(图1),且当初始pH为3.0和4.0时,弱磁场对零价铁去除U(Ⅵ)的促进效果明显高于初始pH为5.0、6.0、7.0的情况。在有弱磁场的条件下,U(Ⅵ)的去除速率随着pH上升而降低;在无弱磁场条件下,U(Ⅵ)的去除速率随着pH上升呈现出先上升后降低的趋势,并在初始pH为4.0时达到最大。为了进一步考察U(Ⅵ)的去除动力学,将反应0~45 min的数据用假一级动力学模型式(1)进行拟合。

式中,obs为假一级动力学反应速率常数,min-1,加弱磁场与无弱磁场条件下拟合所得的速率常数如图1(f)所示。当初始pH为3.0~7.0时,弱磁场使零价铁去除U(Ⅵ)的反应速率常数提高了0.7~11.2倍。无弱磁场时,零价铁去除U(Ⅵ)的假一级动力学常数随着pH的变化相对较小,而当初始pH为3.0~5.0时,有弱磁场情况下的反应速率常数随着pH的升高快速降低。可见在pH较低的情况下,弱磁场的促进效果更为显著。
图1 不同初始pH对零价铁去除U(Ⅵ)动力学的影响及反应0~45 min假一级动力学反应速率常数
Fig.1 Influence of initial pH on U(Ⅵ) removal by ZVI and first-order kinetic rate constants of reaction between 0—45 min
2.2 弱磁场对零价铁去除U(Ⅵ)过程中腐蚀行为的影响
溶液体系中pH、Fe2+浓度的变化在一定程度上可以反映零价铁的腐蚀过程[21],故本研究也检测了零价铁去除U(Ⅵ)过程中pH和Fe2+浓度的变化。如图2所示,随着初始pH的升高,溶液中pH和Fe2+浓度的上升速率呈现出逐渐减缓的趋势,这与零价铁的腐蚀受溶液pH的影响有关。在反应的初始阶段,有弱磁场的情况下,pH和Fe2+浓度的上升速率要高于无弱磁场的情况;随着反应的进行,有弱磁场的情况下,pH和Fe2+的变化呈现出略微下降的趋势,而无弱磁场时pH和Fe2+达到平衡后几乎不变。这是由于在弱磁场作用下,零价铁腐蚀加快,有更多的Fe2+生成,且新生成的Fe2+会与溶液中的氢氧根离子反应生成铁氧化物聚集在零价铁的表面,造成pH和Fe2+浓度的降低。

图2 不同初始pH条件下弱磁场对零价铁去除U(Ⅵ)过程中Fe2+浓度和pH变化的影响
通过扫描电镜可以直观地观察零价铁表面的腐蚀形貌。图3表明,在不同的初始pH条件下,零价铁均发生了部分腐蚀,且随着初始pH的升高,零价铁的腐蚀程度逐渐减小。在初始pH为4.0时,有弱磁场情况下零价铁的腐蚀程度要远高于无弱磁场情况;而随着初始pH的升高(5.0、6.0),有弱磁场情况下的零价铁的腐蚀程度呈现出略高于无弱磁场的趋势。这与弱磁场对零价铁去除U(Ⅵ)动力学(图1)影响的实验现象相吻合。

图3 有弱磁场与无弱磁场条件下零价铁去除U(Ⅵ)腐蚀产物扫描电镜图
综上可知,弱磁场通过促进零价铁的腐蚀促进其对U(Ⅵ)的去除。
2.3 弱磁场促进零价铁去除U(Ⅵ)机理的探究
为了进一步考察腐蚀产物表面铀的价态和铁氧化物的成分,对零价铁去除U(Ⅵ)反应后的固体沉淀进行了XPS光谱分析。根据相关文献[22-23],380.7和391.6 eV为U(Ⅳ)的4f7/2和4f5/2能峰,381.5和392.3 eV为U(Ⅵ)的4f7/2和4f5/2能峰。如图4(a)所示,在不同的反应条件下,U(Ⅵ)只有小部分被还原成四价,而Gu等[8]研究结果表明零价铁腐蚀产物中的铀大部分以U(Ⅳ)形式存在。本研究中铀还原量较少可能是由于实验在敞口搅拌的条件下进行,氧气供应充足,U(Ⅵ)不易被还原成U(Ⅳ)。通过XPS speak fit 分析软件,将U(Ⅳ)与U(Ⅵ)峰面积之比从大到小排列为:pHini5.0,w/WMF>pHini4.0,w/WMF>pHini4.0,w/o WMF>pHini5.0,w/o WMF。在弱磁场的作用下,U(Ⅳ)/U(Ⅵ)的比值要高于无弱磁场的情况。由图1可知,反应3 h后,弱磁场作用下的零价铁腐蚀产物中总铀的含量要高于无弱磁场的情况。这说明了弱磁场不仅能够促进U(Ⅵ)的吸附,同时还促进U(Ⅵ)的还原。当初始pH为4.0时,弱磁场作用下的腐蚀产物中总铀的含量要远高于无弱磁场的情况,这说明了在低pH时,弱磁场的促进效果更为明显。
图4(b)对腐蚀产物表面Fe 2p的XPS光谱进行了分析。参考相关文献[24],α-Fe2O3、Fe3O4的2p3/2能峰分别为710.9 eV、713.8 eV,α-Fe2O3、Fe3O4的2p1/2能峰分别为724.4 eV和724.1 eV,719 eV为α-Fe2O3的卫星扫尾峰。通过分峰拟合,得出Fe3O4与α-Fe2O3峰面积比值从大到小排列为:pHini4.0,w/WMF>pHini4.0,w/o WMF>pHini5.0,w/WMF>pHini5.0,w/o WMF。这说明了在弱磁场的作用下,有更多的Fe3O4的生成。
如图4所示,零价铁腐蚀产物中均存在U(Ⅳ)和U(Ⅵ),且铁氧化物的类别相同,而在弱磁场 的作用下,腐蚀产物中总铀与U(Ⅳ)/U(Ⅵ)的比值都要高于无弱磁场的情况。因此,可以得出的结论是:零价铁去除U(Ⅵ)的主要机理为先吸附,再还原;在反应过程中,弱磁场并不能改变零价铁去除U(Ⅵ)的反应机理,只是加速了其吸附和还原U(Ⅵ)的过程。

图4 外加弱磁场条件下零价铁去除U(Ⅵ)固体产物的光电子能谱分析
2.4 弱磁场对零价铁去除U(Ⅵ)容量的影响
去除容量是影响零价铁去除U(Ⅵ)实际应用的另外一个因素,因此本研究系统地考察了弱磁场对零价铁去除U(Ⅵ)容量的影响。如图5所示,在反应的初始阶段,零价铁对六价铀去除速率较快,反应进行24 h后,零价铁对U(Ⅵ)的去除速率趋于平缓。在去除容量实验中,根据图5,控制反应时间为30 h,达到平衡后计算其去除容量。如图6所示,在U(Ⅵ)初始浓度为1、2 mmol·L-1时,无论是否存在弱磁场,U(Ⅵ)均被零价铁完全去除。随着U(Ⅵ)初始浓度的升高(4、5 mmol·L-1),弱磁场能够明显地提高零价铁对U(Ⅵ)的去除容量,其去除容量为1.7 g·g-1,相比于无弱磁场时,零价铁对U(Ⅵ)的去除容量提高了约0.3倍。

表1 弱磁场强化零价铁去除U(Ⅵ)与其他除U(Ⅵ)技术对比
Note: RT—room temperature.
2.5 弱磁场促进零价铁去除U(Ⅵ)的应用前景
表1总结了相关文献中去除U(Ⅵ)的方法。如表所示,多数文献中报道的用于去除U(Ⅵ)的吸附材料去除容量较小。值得注意的是,Zr1-TiP2O7对U(Ⅵ)的去除容量较大,在接触时间为1 h时,去除容量可达309.8 mg·g-1,但是这种吸附材料制备过程复杂并且耗时,同时吸附材料的稳定性也值得进一步研究。吴唯民等[33]用微生物法实现了含有U(Ⅵ)地下水的原位修复,对于其他技术难以应用的场地治理具有一定的优势。然而微生物法在除U(Ⅵ)过程中对反应条件要求高、操作复杂、反应周期长,限制了其在除铀过程中的应用。纳米零价铁能够实现对U(Ⅵ)的快速去除,但是纳米零价铁存在着潜在的生物毒性,以及较高的成本等缺陷。微米零价铁具有来源广泛、价格低廉等特点被广泛应用。从本研究中可以得出,在弱磁场的作用下,零价铁对U(Ⅵ)具有良好的去除速率和去除容量。相比于其他技术,弱磁场促进零价铁去除U(Ⅵ)具有良好的应用前景。
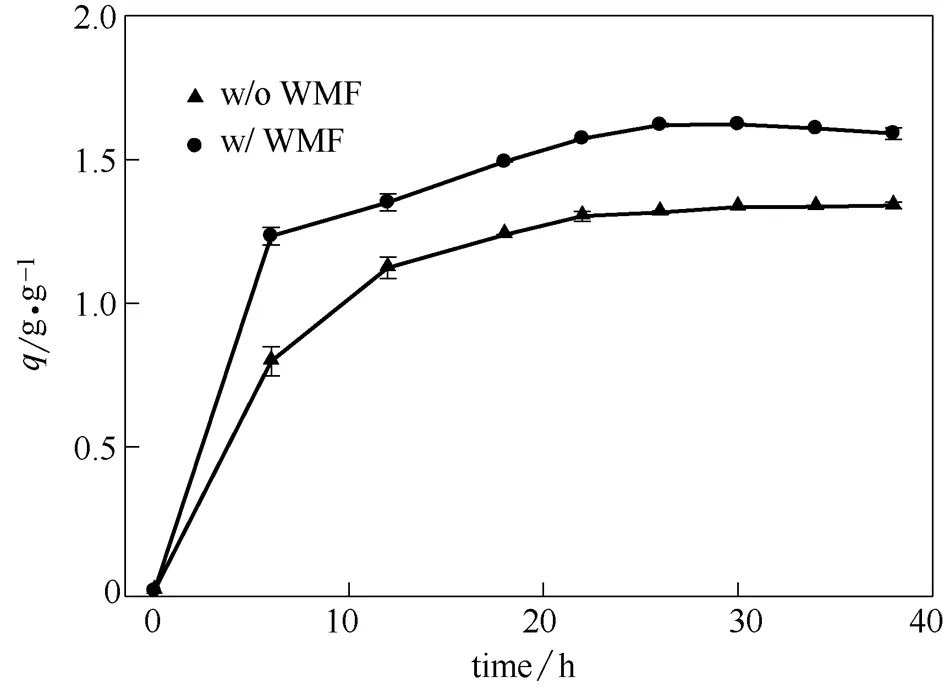
图5 U(Ⅵ)初始浓度为4 mmol·L-1时零价铁去除铀动力学
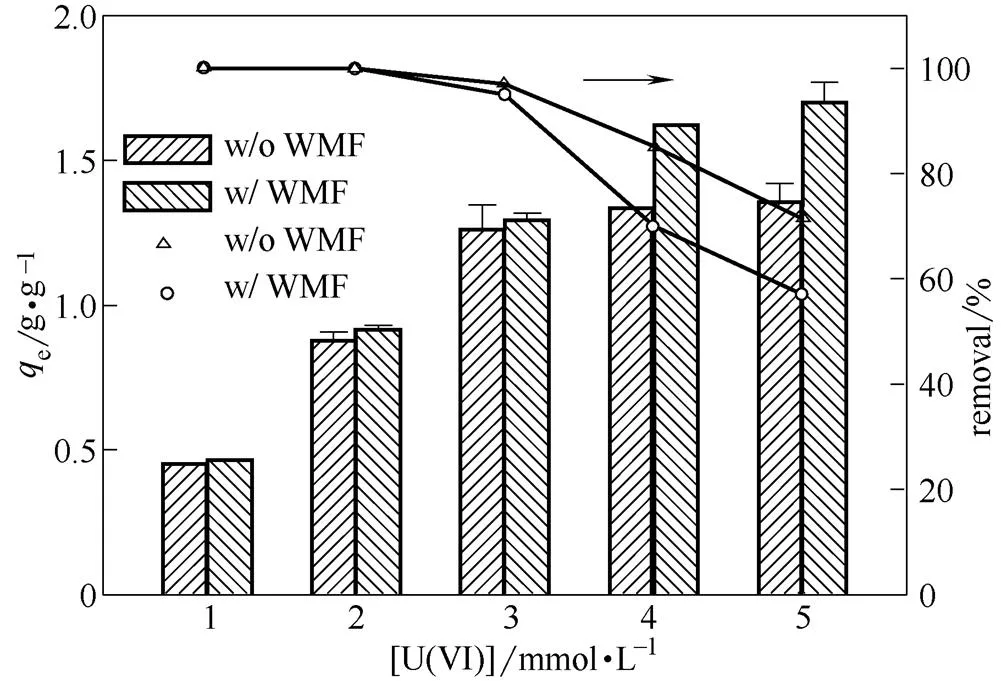
图6不同U(Ⅵ)初始浓度下零价铁对铀的去除容量和去除率
3 结 论
通过外置弱磁场,零价铁对U(Ⅵ)的去除速率和去除容量均有较大提高。在初始pH为3.0的条件下,弱磁场可使零价铁去除六价铀的假一级动力学速率常数提高11.2倍。当初始pH为4.0,零价铁投加量为0.5 g·L-1时,弱磁场作用下的零价铁对U(Ⅵ)的去除容量是无弱磁场时的1.3倍。弱磁场能够促进零价铁的腐蚀,从而加速对U(Ⅵ)的吸附和还原,在此过程中,弱磁场并未改变零价铁去除U(Ⅵ)的机理。相比于其他技术,弱磁场促进零价铁去除U(Ⅵ)具有去除速率快、去除容量高、价格低廉、环境友好、无须额外的能量和药剂投入等特点,因此弱磁场与零价铁协同去除U(Ⅵ)具有良好的应用前景。
References
[1] 何祚庥. 三论我国必须大幅度调整核能政策 [J]. 山东科技大学学报(社会科学版), 2011, 13(5): 1-6. HE Z X. The third relate: we must take adjustments to nuclear policy in our country [J]. Journal of Shandong University of Science and Technology (Social Sciences), 2011, 13(5): 1-6.
[2] 常阳, 崔建勇, 谭靖, 等. 尕斯库勒盐湖水中铀形态分布及影响因素 [J]. 世界核地质科学, 2016, 33: 106-110. CHANG Y, CUI J Y, TAN J,. Analysis on uranium existing forms and its afection factors in saline-water of Gas Hure Lake [J]. World Nuclear Geoscience, 2016, 33: 106-110.
[3] PLACZEK C J, HEIKOOP J M, HOUSE B,. Uranium isotope composition of waters from south Texas uranium ore deposits [J]. Chemical Geology, 2016, 437: 44-55.
[4] 邓冰, 刘宁, 王和义. 铀的毒性研究进展 [J]. 中国辐射卫生, 2010, 19(1): 113-116. DENG B, LIU N, WANG H Y. Uranium toxicity research progress [J]. China Radiation Health, 2010, 19(1): 113-116.
[5] LU C, ZHANG P, JIANG S,Photocatalytic reduction elimination of UO22+pollutant under visible light with metal-free sulfur doped g-C3N4photocatalyst [J]. Applied Catalysis B: Environmental, 2017, 200: 378-385.
[6] GUAN X H, SUN Y K, QIN H J,The limitations of applying zero-valent iron technology in contaminants sequestration and the corresponding countermeasures: the development in zero-valent iron technology in the last two decades (1994—2014) [J]Water Res., 2015, 75: 224-248.
[7] FIEDOR J, BOSTICK W, JARABEK R,. Understanding the mechanism of uranium removal from groundwater by zero-valent iron using X-ray photoelectron spectroscopy [J]Environ. Sci. Technol., 1998, 32: 1466-1473.
[8] GU B, LIANG L, DLCKEY M J,Reductive precipitation of uranium(Ⅵ) by zero-valent iron [J]Environ. Sci. Technol., 1998, 32: 3366-3373.
[9] MATHESON L J, TRATNYEK P G. Reductive dehalogenation of chlorinated methanes by iron metal [J]Environ. Sci. Technol., 1994, 28: 2045-2053.
[10] UHLEMANN M, KRAUSE A, GEBERT AEffects of iron surface pretreatment on kinetics of aqueous nitrate reduction [J]. J. Hazard. Mater., 2005, 126(1/2/3): 189-194.
[11] HUNG H M, HOFFMANN M. Kinetics and mechanism of the enhanced reductive degradation of CCl4by elemental iron in the presence of ultrasound [J]Environ. Sci. Technol., 1998, 32: 3011-3016.
[12] XU J, HAO Z W, XIE C S,Promotion effect of Fe2+and Fe3O4on nitrate reduction using zero-valent iron [J]. Desalination, 2012, 284: 9-13.
[13] HUANG Y H, TANG C L, ZENG H. Removing molybdate from water using a hybridized zero-valent iron/magnetite/Fe(Ⅱ) treatment system [J]Chemical Engineering Journal, 2012, 200/201/202: 257-263.
[14] WANG C B, ZHANG W X. Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs [J]Environ. Sci. Technol., 1997, 31: 2154-2156.
[15] LIEN H L, ZHANG W X. Translation of chlorinated methanes by nanoscale iron particles [J]J. Environ. Eng., 125(11): 1042-1047.
[16] LIANG L P, SUN W, GUAN X H,Weak magnetic field significantly enhances selenite removal kinetics by zero valent iron [J]Water Res., 2014, 49: 371-380.
[17] SUN Y K, GUAN X H, WANG J M.Effect of weak magnetic field on arsenate and arsenite removal from water by zerovalent iron: an XAFS investigation [J]Environ. Sci. Technol., 2014, 48(12): 6850-6858.
[18] FENG P, GUAN X H, SUN Y K.Weak magnetic field accelerates chromate removal by zero-valent iron [J]Journal of Environmental Sciences, 2015, 31: 175-183.
[19] JIANG X, QIAO J L, WANG L.Enhanced paramagnetic Cu2+ions removal by coupling a weak magnetic field with zero valent iron [J]J. Hazard. Mater., 2015, 283: 880-887.
[20] LI J L, BAO H L, XIONG X M,Effective Sb(V) immobilization from water by zero-valent iron with weak magnetic field [J]Separation And Purification Technology, 2015, 151: 276-283.
[21] TRISZCZ J M, PORTA A, EINSCHLAG F S. Effect of operating conditions on iron corrosion rates in zero-valent iron systems for arsenic removal [J]Chemical Engineering Journal, 2009, 150(2/3): 431-439.
[22] ALLEN G C, TRICKLE I R, TUCKER P M. Surface characterization of uranium metal and uranium dioxide using X-ray photoelectron spectroscopy [J]Philosophical Magazine Part B, 2006, 43(4): 689-703.
[23] RIBA O, SCOTT T B, ALLEN G C.Reaction mechanism of uranyl in the presence of zero-valent iron nanoparticles [J]Geochimica et Cosmochimica Acta, 2008, 72(16): 4047-4057.
[24] YAMASHITA T, HAYES P. Analysis of XPS spectra of Fe2+and Fe3+ions in oxide materials [J]Applied Surface Science, 2008, 254(8): 2441-2449.
[25] WANG R, YE J W, RAUT A,Microwave-induced synthesis of pyrophosphate Zr1-TiP2O7and TiP2O7with enhanced sorption capacity for uranium(Ⅵ) [J]J. Hazard. Mater., 2016, 315: 76-85.
[26] 谢水波, 罗景阳, 刘清, 等. 羟乙基纤维素-海藻酸钠复合膜对六价铀的吸附性能及吸附机制 [J]复合材料学报, 2015, 32: 268-275. XIE S B, LUO J Y, LIU Q,AdsorptionofHEC/SA membrane toward U(Ⅵ) and the mechanism [J]. Acta Materiae Compositae Sinica, 2015, 32: 268-275.
[27] JIN Q, SU L, MONTAVON G,. Surface complexation modeling of U(Ⅵ) adsorption on granite at ambient/elevated temperature: Experimental and XPS study [J]Chemical Geology, 2016, 433: 81-91.
[28] ZENG H, SINGH A, BASAK S,. Nanoscale size effects on uranium(Ⅵ) adsorption to hematit [J]Environ. Sci. Technol., 2009, 43: 1373-1378.
[29] KARIMZADEH S N, MERKEL B J. Sorption of uranyland arsenate on SiO2, Al2O3, TiO2and FeOOH [J]Environ. Earth Sci., 2014, 72: 3507-3512.
[30] MAHMOUD M A. Adsorption of U(Ⅵ) ions from aqueous solution using silicon dioxide nanopowder [J/OL]Journal of Saudi Chemical Society, 2016. http://creativecommons.org/licenses/by-nc-nd/4.0/.
[31] SUN Y B, LI J X, WANG X K. The retention of uranium and europium onto sepiolite investigated by macroscopic, spectroscopic and modeling techniques [J]Geochimica et Cosmochimica Acta, 2014, 140: 621–643.
[32] 刘小玲, 陈晓明, 宋收, 等. 柠檬酸杆菌对U(Ⅵ)的去除效应及机理研究 [J]核农学报, 2015, 29: 1774-1781. LIU X L, CHEN X M, SONG S,The effect and mechanism oftoward U(Ⅵ) removal [J]. Journal of Nuclear Agricultural Sciences, 2015, 29: 1774-1781.
[33] 吴唯民, JACK W, DAVID W. 地下水铀污染的原位微生物还原与固定:在美国能源部田纳西橡树岭放射物污染现场的试验 [J]环境科学学报, 2011, 31: 449-459. WU W M, JACK W, DAVID W. Bio reduction and immobilization of uranium insitu: a case study at a USA department of energy radioactive waste site, OakRidge, Tennessee [J]. Journal of Environmental Science, 2011, 31: 449-459.
[34] SHENG G D, YANG P J, TANG Y N,New insights into the primary roles of diatomite in the enhanced sequestration ofby zerovalent iron nanoparticles: an advanced approach utilizing XPS and EXAFS [J]Applied Catalysis B: Environmental, 2016, 193: 189-197.
[35] YAN S, CHEN Y H, XIANG W,Uranium (Ⅵ) reduction by nanoscale zero-valent iron in anoxic batch systems: the role of Fe(Ⅱ) and Fe(Ⅲ) [J]Chemosphere, 2014, 117: 625-630.
Enhancing reactivity of zerovalent iron toward U(Ⅵ) by weak magnetic field
CAO Bei, LI Jinxiang, GUAN Xiaohong
(College of Environment Science and Engineering, Tongji University, Shanghai 200092, China)
The influence of weak magnetic field (WMF) on the process of U(Ⅵ) removing byzero valent iron (ZVI) was explored and the main mechanism was investigated. The weak magnetic field could obviously promote the U(Ⅵ) sequestration under the condition of different initial pH (pHini). The first-order kinetic rate constants of U(Ⅵ) removal by ZVI with WMF at pH 3.0—7.0 were about 0.7 to 11.2 fold greater than those without WMF. The removal capacity of zero-valent iron toward U(Ⅵ) was 1.7 g·g-1with WMF at initial pH 4.0 and Fe 0.5 g·L-1, being of 0.3-fold higher than that without WMF. The weak magnetic field could promote the corrosion of the zero-valent iron, thereby increasing its removal of U(Ⅵ), which could be verified by SEM, [Fe2+] and pH variation. The main mechanism of U(Ⅵ) removal by zero valent iron was adsorption together with reduction. The application of WMF did not change the mechanisms but accelerated its adsorption and reduction toward U(Ⅵ). As a chemical-, energy-free and environmental-friendly method, improving the reactivity of ZVI by WMF superimposition was novel and promising in the prospect of U(Ⅵ) sequestration from water.
weak magnetic field; zero-valent iron; U(Ⅵ); kinetics; corrosion; adsorption; removal capacity
10.11949/j.issn.0438-1157.20170196
O 614.81+1
A
0438—1157(2017)08—3282—09
关小红。第一作者:曹贝(1991—),男,硕士研究生。
国家自然科学基金项目(21522704)。
2017-02-28收到初稿,2017-04-25收到修改稿。
2017-02-28.
Prof.GUAN Xiaohong, guanxh@tongji.edu.cn
supported by the National Natural Science Foundation of China (21522704).
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
当代陕西(2022年6期)2022-04-19
娃娃乐园·综合智能(2021年12期)2022-01-18
粉末冶金技术(2021年3期)2021-07-28
当代化工(2020年8期)2020-09-09
天然产物研究与开发(2019年10期)2019-11-05
化工管理(2017年12期)2017-05-12
中学生数理化·高二版(2016年12期)2017-02-28
表面工程与再制造(2016年5期)2016-12-15
中国民族医药杂志(2016年2期)2016-05-14
