
美洲大蠊基因组重复序列分析
2017-10-12 10:51牟必琴严超超李午佼李静沈咏梅岳碧松
四川动物 2017年5期
牟必琴, 严超超, 李午佼, 李静, 沈咏梅, 岳碧松*
(1.四川大学生命科学学院,生物资源与生态环境教育部重点实验室,成都610064; 2. 药用美洲大蠊四川省重点实验室,成都610081)
美洲大蠊基因组重复序列分析
牟必琴1, 严超超1, 李午佼1, 李静1, 沈咏梅2, 岳碧松1*
(1.四川大学生命科学学院,生物资源与生态环境教育部重点实验室,成都610064; 2. 药用美洲大蠊四川省重点实验室,成都610081)
重复序列是真核生物基因组的重要组成部分。一些重复序列,如自主型的逆转录转座子LINE,在昆虫的系统进化和遗传多样性研究方面得到了广泛的应用。denovo从头预测和基于同源比对预测相结合的方法被用来搜索美洲大蠊Periplanetaamericana基因组,共鉴定出大约占全基因组62%的重复序列。研究发现,散在重复序列中,DNA转座子占美洲大蠊基因组的16.18%;逆转座元件中LINE最多,占基因组的13.64%,SINE和LTR分别占基因组的3.52%和1.32%。LINEs中的BovBs亚家族在所有转座子亚家族中比例最高(约6.73%)。美洲大蠊与德国小蠊Blattellagermanica相比,除LTR外,其他类型的转座子占基因组的比例均高于德国小蠊。通过分析逆转录转座子反转录酶完整度、氨基酸序列相似度及遗传距离,从美洲大蠊基因组中鉴定出一类BovBs:RTE-1_PAm。BovBs的反转录酶氨基酸序列的系统树表明,美洲大蠊与内华达古白蚁Zootermopsisnevadensis的进化关系比与其同属蜚蠊科Blattidae的德国小蠊的关系更近。昆虫中BovBs的进化关系与传统核基因进化关系的不同,表明转座子的进化相对宿主基因的进化具有一定的独立性。
美洲大蠊; 基因组; 重复序列; 转座子; BovBs; 系统进化
Abstract: Repetitive sequences constitute a large fraction of a eukaryote genome. Some classes of repetitive sequences, such as LINEs, which are a member of autonomous transposons, have been widely applied in molecular phylogenetic and genetic diversity studies of insects. In this study,denovoprediction and homology alignment were used to search the repetitive sequences inPeriplanetaamericanagenome, and the results showed that the screened repetitive sequences accounted for approximately 62% of the genome. Among interspersed repetitive sequences, the DNA transposons constituted 16.18% of the whole genome. In retrotransposons, LINEs accounted for 13.64% of the genome followed by SINE (3.52%) and LTR (1.32%), respectively. The BovBs subfamily in LINEs was the most abundant (6.73%) compared to other classes of transposons. The proportion of all classes of transposable elements inP.americanawere higher than that ofBlattellagermanicaexcept the LTRs. According to the integrality of the reverse transcriptase of retrotransposons, similarities and genetic distance of the amino acid sequences, a kind of BovB retrotransposons were identified inP.americanagenome and named RTE-1_PAm. Phylogenetic analyses based on the amino acid sequences of BovBs reverse transcriptase indicated thatP.americanashared closer relationship withZootermopsisnevadensisthan withB.germanicawhich are both Blattidae. The difference of phylogenetic analyses results among these insects inferred by using retrotransposons and karyogene suggested an independent evolutional history of TEs relative to the host karyogene.
Keywords:Periplanetaamericana; genome; repetitive sequences; transposable element; BovB retrotransposon; phylogeny
重复序列是真核基因组的重要组成部分,它分为串联重复序列和散在重复序列,其中转座子是散在重复序列的主要组成部分。转座子又称为转座元件(transposable element,TE),是广泛分布于真核生物基因组中的一种可移动的DNA序列(Kidwell & Lisch,2002),是宿主进化的重要动力之一(Kazazian,2004)。按转座机制和序列特征,散在重复序列可分为DNA转座子和逆转录转座子两大类。逆转录转座子编码逆转录酶,该酶由DNA转录形成的RNA经逆转录转换成cDNA,最后将形成的cDNA整合到基因组完成转座,引起稳定突变(Eickbushetal.,2002)。根据序列两端是否含有长末端重复序列,逆转录转座子又分为长末端重复序列(long terminal repeat, LTR)和非长末端重复序列。非自主的非长末端重复序列称为短散在重复序列(short interspersed nuclear element, SINE)。非自主重复序列不能独立转座,自身不编码有功能的逆转录转座酶,只能依赖于相关的自主元件进行转座。相反,自主的非长末端重复序列称为长散在重复序列(long interspersed nuclear element, LINE),它本身含有有功能的反转录酶而完成自身的转座。DNA转座子编码转座酶,一般直接通过“剪切和粘贴”机制发生转座,引起不稳定突变(Ohshimaetal.,1996)。
关于昆虫BovB逆转录转座子(BovB retrotransposons, BovBs)的进化研究迄今较少。BovBs属于LINE,全长约3.2 kb,在蜚蠊目Blattodea物种的基因组中均占很高比例。完整的BovBs中含有一个长约1 000个氨基酸的开放阅读框,用于编码逆转录转座子转座活动过程中需要的4种酶:反转录酶、核酸内切酶、核酸外切酶及磷酸酶(Malik & Eickbush,1998)。BovBs在各类真核生物的基因组中均有报道,比如在短尾负鼠Monodelphisdomestica、牛、蜥形类和猛犸象Mammuthusprimigenius等物种的基因组中被发现占很高比例(Gentlesetal.,2007;Adelsonetal.,2009;Kordis,2009;Zhao & Qi,2009)。近年来,昆虫中的BovBs也陆续被发现,如家蚕Bombyxmori(Mortonetal.,2012)和肩突硬蜱Ixodesscapularis(Tayetal.,2010)。此外,一些研究对蝇蛹金小峰Nasoniavitripennis、红带袖蝶Heliconiusmelpomene、果蝇属Drosophila、埃及伊蚊Aedesaegypti、致倦库蚊Culexquinquefasciatus和按蚊属Anopheles基因组中BovBs进行了结构和活性的相关研究(Clark & Eisen,2007;Sinkins,2007;Arensburgeretal.,2010;Webbetal.,2011;Consortiumetal.,2012)。
随着二代测序技术的飞速发展,越来越多物种的全基因组得以揭示,在物种的全基因组中鉴定重复序列的研究也越来越多。美洲大蠊Periplanetaamericana属于蜚蠊目蜚蠊科Blattidae大蠊属Periplaneta,具有较强的抗逆和适应能力,是世界性的卫生害虫,同时在中国也是重要的药用昆虫。蜚蠊目的德国小蠊Blattellagermanica为其同目同科物种。近年来,原等翅目Blattaria的内华达古白蚁Zootermopsisnevadensis被归类于蜚蠊目(Terraponetal.,2014)。为了了解美洲大蠊的遗传背景,本研究利用denovo预测和同源性搜索相结合的方法首次对美洲大蠊基因组重复序列进行鉴定,并比较分析了重复序列在美洲大蠊、德国小蠊和内华达古白蚁3种蜚蠊目昆虫基因组中的分布规律,最后以BovBs内反转录酶编码序列作为分子标记,初步研究了BovBs在16种昆虫中的进化关系。
1 基因组来源与方法
1.1基因组来源
美洲大蠊样品来自四川好医生攀西药业有限公司西昌饲养基地。使用二代Illumina和三代Pacbio测序相结合的方法,分别得到了460×和10×的原始数据。通过DISCOVAR (https://www.broad-institute.org/software/discovar/blog)(Loveetal.,2016)和BESST(Sahlinetal.,2014,2016)组装得到美洲大蠊全基因组。德国小蠊和内华达古白蚁的基因组均下载自NCBI (PRJNA203136和PRJNA203242)。
1.2方法
1.2.1重复序列的鉴定使用3种方案搜集预测基因组中的重复序列:(1)从头预测转座子:使用RepeatModeler(http://www.repeatmasker.org,version 1.0.8)扫描基因组序列,对不同重复序列的特性进行计算机建模,得到非冗余的重复序列数据库(Smit & Hubley,2008-2015),然后使用RepeatMasker(http://www.repeatmasker.org,version 4.0.5)进行重复序列的搜索和识别(Smitetal.,2013-2015);(2)基于同源比对注释重复序列:使用RepeatMasker和RepBase重复序列数据库(version 19.09)进行比对并注释(Jurkaetal.,2005;Baoetal.,2015);(3)使用RepeatProtein(version 4.0.5)和它的默认参数搜索已知转座子内的蛋白质序列相关的重复序列(Huetal.,2012)。
1.2.2重复序列的统计重复序列统计的原始文件为RepeatMasker的输出文件(“.out”)和RepeatProtein的输出文件(“.annot”)。在对重复序列统计之前,首先对每种类型的重复序列过滤,去掉核苷酸序列长度小于10 bp和与重复序列库相似度小于70%的重复元件。这一过程使用shell和python编写的本地脚本完成。过滤得到的文件使用R语言统计每个物种基因组中重复序列的种类、占基因组的比例并作图。
1.2.3BovBs进化分析使用LINE保守的反转录酶区域的氨基酸序列构建昆虫的系统进化树。首先,将参考基因组的BovBs序列作为质询序列,使用Blastx搜索出与NCBI的蛋白保守序列库(CDD)中LINE中的反转录酶氨基酸保守序列(cd01650)同源性较高的蛋白质序列(evalue值不大于1e-10,氨基酸长度大于100 aa)(Sun & Mueller,2014);然后将所得的蛋白质序列进行修整(如去除低质量的插入缺失位点),保留CDD库内的一致性序列,然后使用CD-HIT去除冗余序列(Huangetal.,2010),得到氨基酸序列;最后使用RepeatMasker中的COSEG(Lavoieetal.,2013)合并得到BovBs中氨基酸的一致性序列。使用MEGA 5.2(Tamuraetal.,2011;Sormachevaetal.,2012)计算氨基酸序列之间的遗传距离。此外,从RepBase重复序列蛋白质序列库中下载了其他13种昆虫的19条BovBs蛋白质序列(表1)。使用ClustalW (Thompsonetal.,1999)和prank v.140603 (Löytynoja & Goldman,2010)将氨基酸序列集进行对齐。最后使用MEGA 5.2(Tamuraetal.,2011)构建最大似然树,Bootstrap设置为1 000以检验分子系统树各分支的置信度,并使用MrBayes 3.2(Ronquist & Huelsenbeck,2003)构建贝叶斯树。
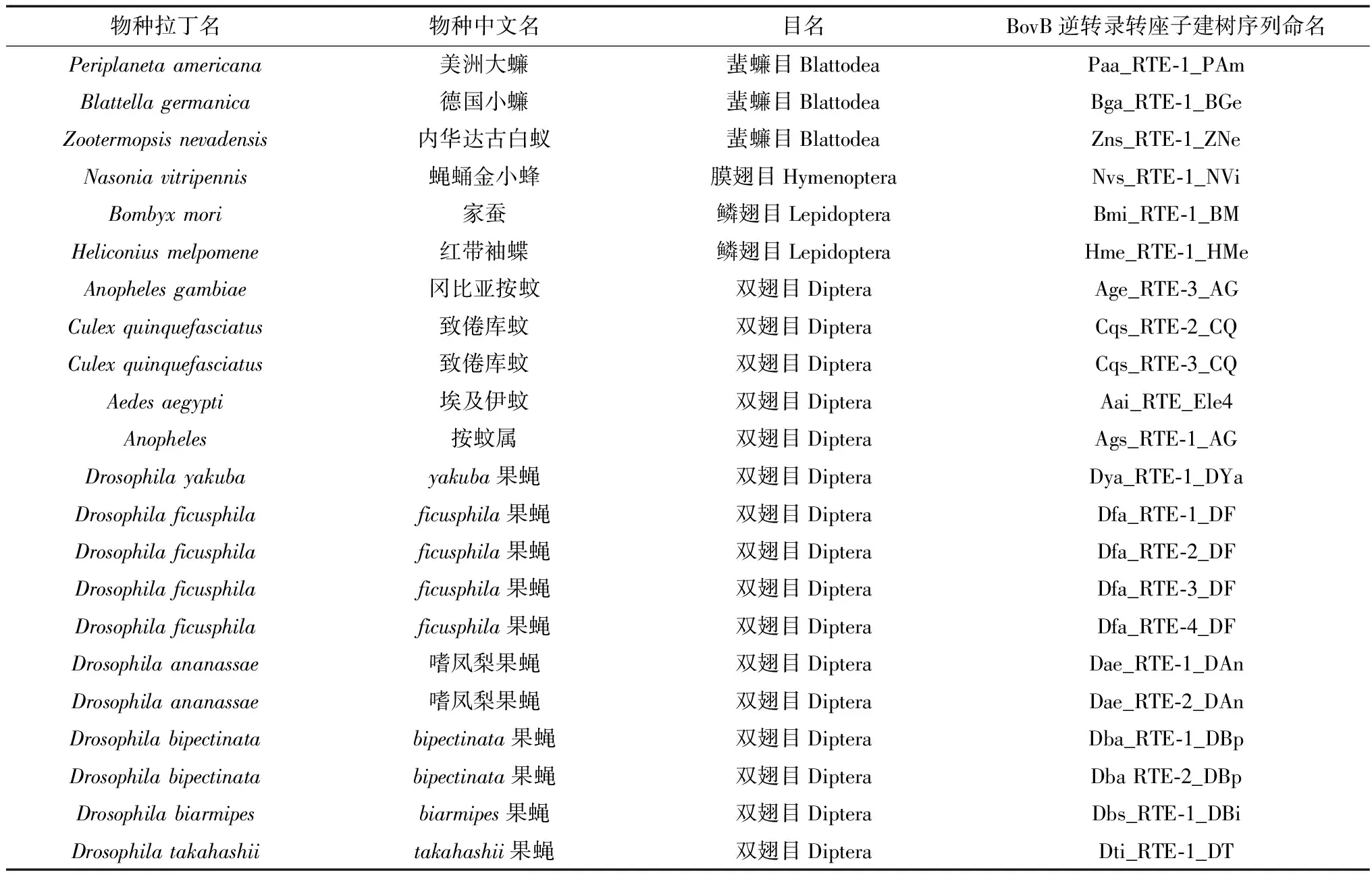
表1 BovB逆转录转座子氨基酸序列信息Table 1 The amino acid sequence information of BovB retrotransposons
注: BovBs序列名称的命名规则为: 物种拉丁名属名的首字母和种名首尾字母+BovB逆转录转座子的亚家族名。
Notes: The sequence names consists of the first letter of genus name, the first and the end letters of species’ name, as well as the subfamily name of BovB retrotransposons.
2 结果
2.1主要重复序列和比例
在美洲大蠊基因组中,使用RepeatModelerdenovo预测方法贡献最大,得到了1 862.3 Mp的重复序列,占基因组的58.27%;使用RepBase同源比对注释得到了355.3 Mp的重复序列,占基因组的11.12%;使用RepeatProtein识别的转座子蛋白序列库得到了417.5 Mp的重复序列,占基因组的13.06%。综合以上3种不同的方法,去除冗余后(不同方法可能在同一个位置预测出转座子),共得到了1 993.9 Mp的非冗余重复序列,占基因组的62.38%(图1,表2),这和人类基因组的重复序列占比相似(International Human Genome Sequencing Consortium,2001)。在美洲大蠊基因组重复序列中,未知类型的重复序列(Unknown)占总重复序列的1/3左右,占基因组的25.75%。已知类型转座子中,DNA转座子总长度占基因组的16.18%,相对于所有的逆转录转座子(占基因组的18.75%)较小。其中,LINE为13.64%。此外,SINE和LTR比例均较少,只占1.80%左右。其他类型的重复序列,如卫星重复序列、核糖体重复序列及其他简单重复序列占基因组的3.70%(图1,表2)。
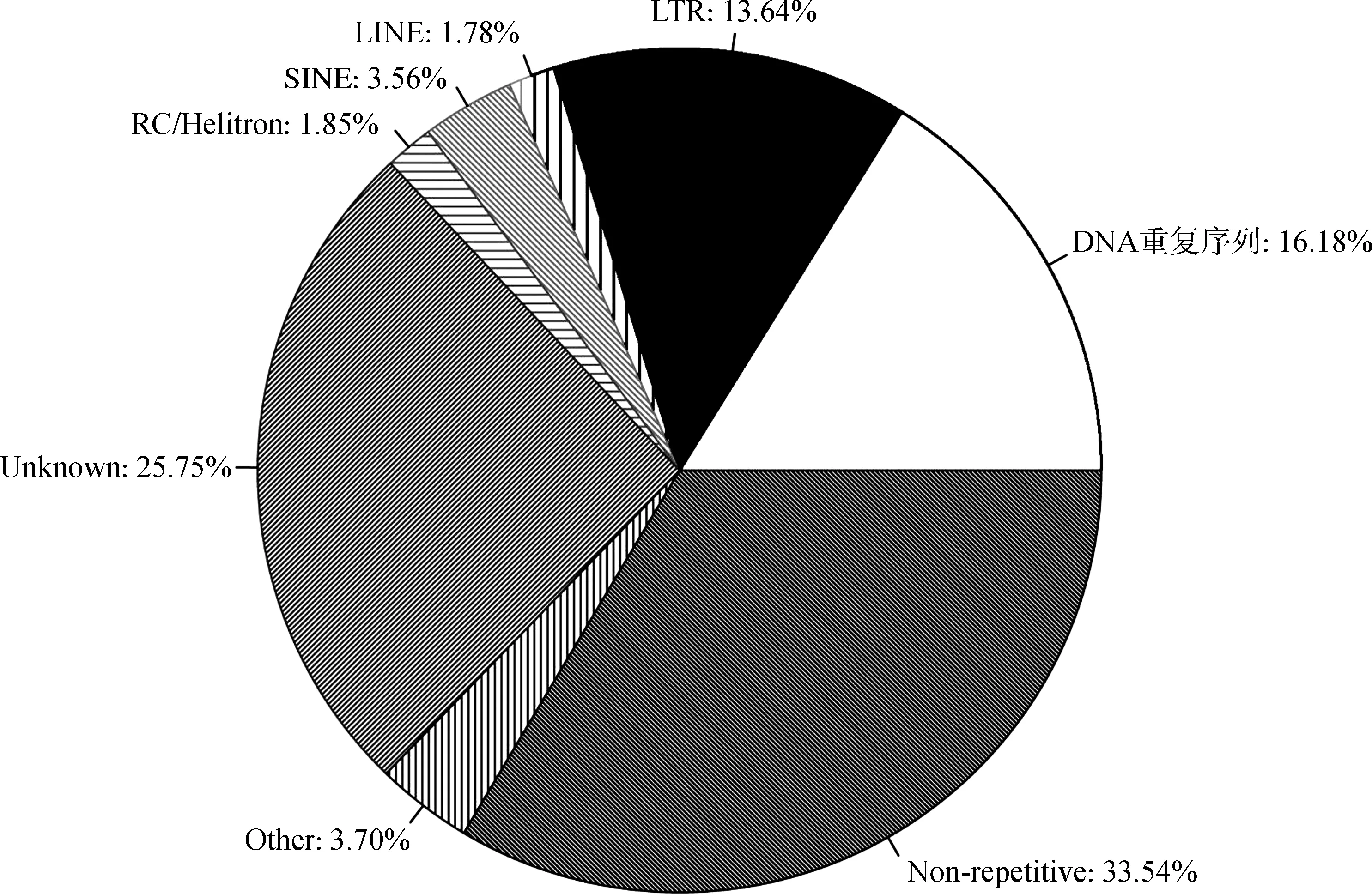
图1 美洲大蠊基因组中重复序列分布图Fig. 1 The pie distribution diagram of the repetitive sequences in the Periplaneta americana genome

重复序列种类ClassesofrepeatsRepeatModeler注释RepeatModelerRepBase库注释RepBaseRepeatProtein库注释RepeatProtein重复序列总和Total长度Length/Mp占基因组比例Percentageofgenome/%长度Length/Mp占基因组比例Percentageofgenome/%长度Length/Mp占基因组比例Percentageofgenome/%长度Length/Mp占基因组比例Percentageofgenome/%DNA442413841129353141944451701618LINE327110231238387268584043601364SINE112335295030001139356LTR42213213304257018570178RC/Helitron5761804101318006591185Other868272998312001182370Unknown82222572090030082312575Total1862358273553111241751306199396238
蜚蠊目物种不同类型的重复序列比较分析结果见表3。美洲大蠊基因组的重复序列含量(62.38%)略高于德国小蠊(61.94%),远高于内华达古白蚁(35.84%)。这一结果与脊椎动物中随基因组的增加重复序列占基因组比例增大的趋势一致(Chalopinetal.,2014;Chalopinetal.,2015)。DNA型重复序列是美洲大蠊和德国小蠊基因组中最丰富的重复序列类型,其中美洲大蠊的DNA型重复序列含量最高,达16.18%,德国小蠊次之(13.07%),均显著高于内华达古白蚁(6.20%)。同属于DNA重复序列的RC/Helitron型重复序列与这一现象有所不同:美洲大蠊最高,德国小蠊最低。而LINE是内华达古白蚁基因组中最丰富的重复序列类型,约占基因组10%,但这一比例仍然低于美洲大蠊(13.64%)和德国小蠊(12.13%)。SINE在美洲大蠊和德国小蠊中的含量大致相当(大约3%),均高于内华达古白蚁(2.24%)。虽然大多数转座子类型在美洲大蠊基因组中都超过德国小蠊,但德国小蠊基因组中LTR转座子(5.02%)的比例却显著高于美洲大蠊(1.78%)。
2.2主要重复序列亚家族比较分析
DNA转座子亚家族(TcMar、hAT)和LINE逆转录转座子亚家族(BovB、L2)为3个物种基因组中重复序列的最主要组成部分(图2),尤其是BovBs在3个近缘物种中均占有较高比例,美洲大蠊的BovBs也高于其他2个物种。而LTR逆转录转座子亚家族中,德国小蠊的Copia、Gypsy和Pao亚家族比例高于美洲大蠊。相反,美洲大蠊的SINE类型的ID亚家族(0.85%)比例明显高于德国小蠊(0.00%)。除此之外,内华达古白蚁的BovBs在其基因组中的比例最高(5.92%),且高于德国小蠊(4.50%),这归功于BovBs属于新型转座子类型的LINE逆转座子。
2.3美洲大蠊BovBs的序列分析
由于BovBs在美洲大蠊、德国小蠊及内华达古白蚁中占基因组比例均很高,可被用于转座子系统进化研究。将BovBs通过Blastx比对到反转录酶氨基酸序列库,分别得到3个物种具有90%以上同源性的一系列氨基酸序列。使用COSEG得到它们的一致性序列并分别命名为RTE-1_PAm,RTE-1_BGe和RTE-1_ZNe。将它们与其他13种昆虫的19条BovBs氨基酸序列一起进行比较分析。本研究的蜚蠊目昆虫和部分其他目昆虫的遗传距离及相似性结果见表4,蜚蠊目和与蜚蠊目相关的其他昆虫BovBs反转录酶的遗传距离为0.29~1.14,序列的相似度34.30%~75.20%。其中美洲大蠊与内华达古白蚁的反转录酶具有最高的相似度,为75.20%,与之对应,两者遗传距离最小,为0.29。美洲大蠊与德国小蠊的反转录酶的相似度为65.20%,它们的遗传距离值为0.43,明显大于美洲大蠊与内华达古白蚁的遗传距离。美洲大蠊与红带袖蝶的BovBs反转录酶同源性最低,相似度为34.30%,遗传距离值为1.07。
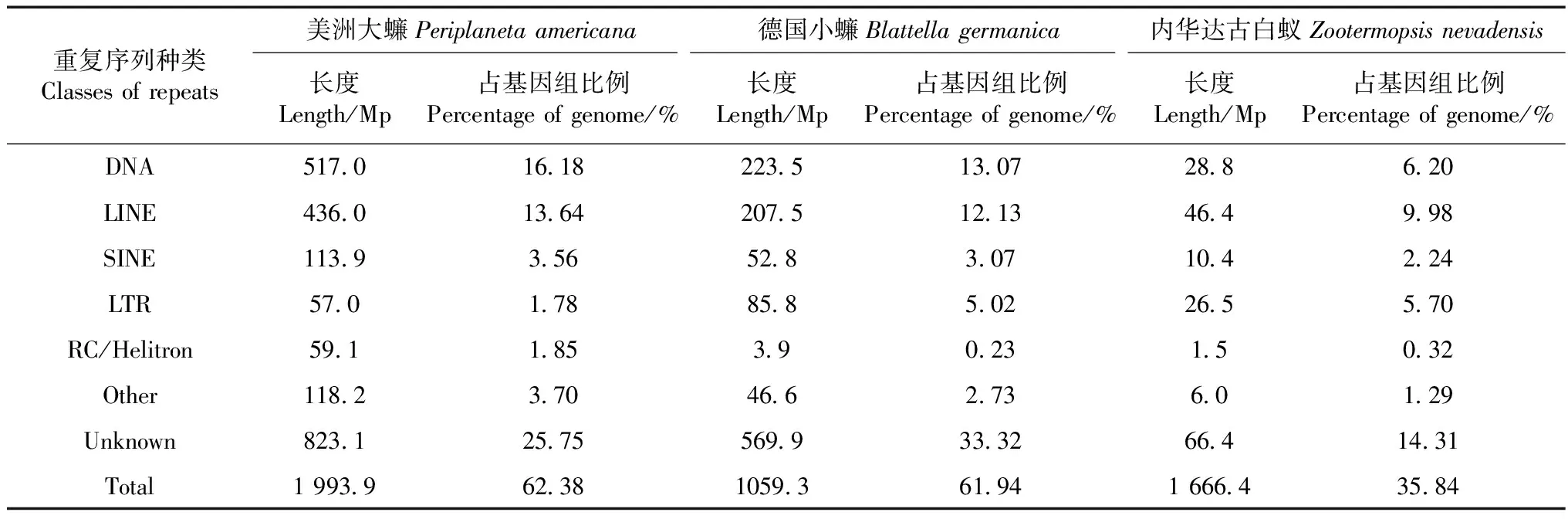
表3 不同物种重复序列的比较Table 3 Comparison of repetitive sequences in different species

图2 3种蜚蠊目昆虫基因组主要重复序列亚家族比较分析Fig. 2 Comparative analysis of the major repetitive sequence subfamilies in 3 Blattodea insect genomes
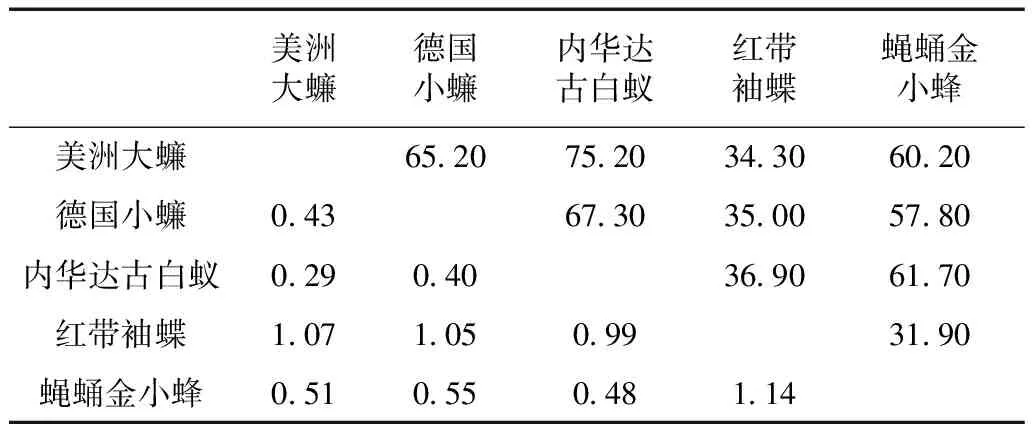
表4 部分昆虫BovB逆转录转座子氨基酸序列的遗传距离及相似性(%)Table 4 The genetic distance and similarity (%) ofBovB retrotransposons in some insect genomes
注: 对角线上半部分数据为相似度, 下半部分数据为遗传距离。
Notes: The data above the diagonal are the similarities between BovB retrotransposons, while those below the diagonal are the genetic distance.
2.4BovBs系统进化分析
将RTE-1_PAm,RTE-1_BGe和RTE-1_ZNe与其他昆虫的19条BovBs家族的氨基酸进行序列比对,采用贝叶斯法和最大似然法构建分子系统发育树。来自4个目16个物种的22条氨基酸序列被分为4大类群(图3)。类群Ⅰ包含鳞翅目的家蚕和红带袖蝶。类群Ⅱ和Ⅲ均属于双翅目,类群Ⅱ含有双翅目的果蝇属物种,与鳞翅目聚在一起。类群Ⅲ既含有果蝇类,又含有蚊类,其中按蚊属与果蝇属聚在一个小支上。其他3种蚊类聚在另外一个小支上,包括1种埃及伊蚊、2条致倦库蚊和1种冈比亚按蚊。类群Ⅳ中包括膜翅目的蝇蛹金小蜂和蜚蠊目的美洲大蠊、德国小蠊以及内华达古白蚁,类群Ⅳ位于系统进化树内部,证明本研究鉴定的蜚蠊目的BovBs非常可靠。美洲大蠊和内华达古白蚁的BovBs聚在同一个进化小支上,且两者的进化距离较德国小蠊与美洲大蠊的距离更近(图3,表4)。
3 讨论
在DNA转座子类型中,2种蜚蠊科昆虫的DNA转座子占基因组的比例均远高于内华达古白蚁,这种差异主要表现在2种蜚蠊科昆虫DNA转座子中的2个亚家族hAT和TcMar占基因组的比例明显高于内华达古白蚁。在已知的反转录转座子类型中,美洲大蠊LTR的类型和占基因组的比例明显低于德国小蠊,但其他转座子类型均是美洲大蠊略高于德国小蠊。从总体上看,美洲大蠊、德国小蠊和内华达古白蚁重复序列占基因组比例呈递减趋势,这与它们基因组大小依次递减一致,符合随着基因组的增大,重复序列占基因组比例也增大的规律(Chalopinetal.,2014,2015)。美洲大蠊重复序列最多的类型为Unknown(25.75%),说明其基因组中未知的重复序列类型还有很多,在这些未知类型的重复序列中除一部分简单重复序列外,还可能存在一些物种特异的新类型重复序列,有待进一步挖掘分析。
采用存在于昆虫基因组中且某些物种含量丰富的BovBs进行比较研究和系统进化分析,为理解其在昆虫基因组的组成和进化历程奠定了一定的基础。美洲大蠊、德国小蠊和内华达古白蚁3个物种基因组中的一类BovBs序列,它们的氨基酸同源性高达90%。比对发现,RTE-1_PAm与RTE-1_BGe相似度为65.20%,而与RTE-1_ZNe聚在同一支上且其相似度为75.20%,与传统的分类和系统进化关系(德国小蠊与美洲大蠊关系较近)不同(Misofetal.,2014)。形成这种差异的原因可能是转座子的进化速率与基因组不一致,即转座子的同义替换速率要低于或高于宿主基因的同义替换速率(Sánchezgraciaetal.,2005)。除此之外,选择压力和修复机制的不同也有可能使转座子与宿主基因的进化速率出现差异。要解决这一问题,需要对更多昆虫基因组的特性和转座子的进化历程进行更深入的研究。
转座子在基因组中随意跳动,导致基因组序列的删除、扩增、移位、断裂和重组等现象(Langleyetal.,1988;Charlesworthetal.,1994),从而影响物种的正常生命活动(Le & Capy,2006;Oliver & Greene,2009)。近年来,昆虫中重复序列的研究较少,已有的研究主要集中于鳞翅目和双翅目的果蝇属中。例如家蚕基因组中的MITEs、Chapaev、Tcl/Mariner等转座子的转座活动使基因组发生重组(Zhangetal.,2014)。果蝇属的P转座子的扩增和水平转移影响了基因组的变化(Clark & Kidwell,1997;Arcà & Savakis,2000)。本研究首次对美洲大蠊基因组的重复序列进行了系统的统计描述,并结合近缘物种的重复序列进行比较分析,这为美洲大蠊基因组研究提供了重要支持,对昆虫重复序列的研究具有积极作用。

图3 BovB逆转录转座子反转录酶氨基酸序列系统进化树Fig. 3 Phylogenetic relationship among BovB retrotransposons based on the amino acid sequence of reverse transcriptase
Adelson DL, Raison JM, Edgar RC. 2009. Characterization and distribution of retrotransposons and simple sequence repeats in the bovine genome[J]. Proceedings of the National Academy of Sciences, 106(31): 12855-12860.
Arcà B, Savakis C. 2000. Distribution of the transposable element Minos in the genusDrosophila[J]. Genetica, 108(3): 263.
Arensburger P, Megy K, Waterhouse RM,etal. 2010. Sequencing ofCulexquinquefasciatusestablishes a platform for mosquito comparative genomics[J]. Science, 330(6000): 86-88.Bao W, Kojima KK, Kohany O. 2015.Repbase update, a database of repetitive elements in eukaryotic genomes[J]. Mobile DNA, 6(1): 11.
Chalopin D, Fan S, Simakov O,etal. 2014. Evolutionary active transposable elements in the genome of the coelacanth[J]. Journal of Experimental Zoology Part B Molecular & Developmental Evolution, 322(6): 322-333.
Chalopin D, Naville M, Plard F,etal. 2015. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates[J]. Genome Biology Evolution, 7(2): 567-580.
Charlesworth B, Sniegowski P, Stephan W. 1994. The evolutionary dynamics of repetitive DNA in eukaryotes[J]. Nature, 371(6494): 215-220.
Clark AG, Eisen MB. 2007. Evolution of genes and genomes on theDrosophilaphylogeny[J]. Nature, 450(7167): 203-218.
Clark JB, Kidwell MG. 1997. A phylogenetic perspective on P transposable element evolution inDrosophila[J]. Proceedings of the National Academy of Sciences of the United States of America, 94(21): 11428.
Consortium THG, Dasmahapatra KK, Walters C,etal. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species[J]. Nature, 487(7405): 94-98.
Eickbush TH, Malik HS, Eickbush TH. 2002. Origins and evolution of retrotransposons[J]. Mobile DNA: 1111-1144.
Gentles AJ, Wakefield MJ, Kohany O,etal. 2007. Evolutionary dynamics of transposable elements in the short-tailed opossumMonodelphisdomestica[J]. Genome Research, 17(7): 992-1004.
Hu Q, Ma T, Wang K,etal. 2012. The yak genome database an integrative database for studying yak biology and high-altitude adaption[J]. BMC Genomics, 13(2): 600.
Huang Y, Niu B, Gao Y,etal. 2010. CD-HIT suite: a web server for clustering and comparing biological sequences[J]. Bioinformatics, 26(5): 680-682.
International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome[J]. Nature, 409: 860-921.
Jurka J, Kapitonov VV, Pavlicek A,etal. 2005. Repbase update, a database of eukaryotic repetitive elements[J]. Cytogenetic & Genome Research, 110(1-4): 462.
Kazazian HH. 2004. Mobile elements: drivers of genome evolution[J]. Science, 303(5664): 1626-1632.
Kidwell M, Lisch D. 2002.Transposable elements as sources of genomic variation[M]// Craig NL, Cralgie R, Gellert M,etal. Mobile DNA II. Washington DC: ASM Press: 59-90.Kordis D. 2009. Transposable elements in reptilian and avian (sauropsida) genomes[J]. Cytogenetic & Genome Research, 127(2-4): 94-111.
Langley CH, Montgomery E, Hudson R,etal. 1988. On the role of unequal exchange in the containment of transposable element copy number[J]. Genetics Research, 52(3): 223-235.
Lavoie CA, Platt RN, Novick PA,etal. 2013. Transposable element evolution inHeliconius, suggests genome diversity within Lepidoptera[J]. Mobile DNA, 4(1): 21.
Le RA, Capy P. 2006. Population genetics models of competition between transposable element subfamilies[J]. Genetics, 174(2): 785-793.
Love RR, Weisenfeld NI, Jaffe DB,etal. 2016. Evaluation of DISCOVARdenovo, using a mosquito sample for cost-effective short-read genome assembly[J]. BMC Genomics, 17(1): 1-10.
Löytynoja A, Goldman N. 2010. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser[J]. BMC Bioinformatics, 11(1): 1-6.
Malik HS, Eickbush TH. 1998. The RTE class of non-LTR retrotransposons is widely distributed in animals and is the origin of many SINEs[J]. Molecular Biology and Evolution, 15(9): 1123-1134.
Misof B, Liu S, Meusemann K,etal. 2014. Phylogenomics resolves the timing and pattern of insect evolution[J]. Science, 346(346): 763-767.
Morton WA, Kortschak RD, Gardner MG,etal. 2012. Widespread horizontal transfer of retrotransposons[J]. Proceedings of the National Academy of Sciences of the United States of America, 110(3): 1012-1016.
Ohshima K, Hamada M, Terai Y,etal. 1996. The 3’ ends of tRNA-derived short interspersed repetitive elements are derived from the 3’ ends of long interspersed repetitive elements[J]. Molecular & Cellular Biology, 16(7): 3756-3764.
Oliver KR, Greene WK. 2009. Transposable elements: powerful facilitators of evolution[J]. Bioessays, 31(7): 703-714.
Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models[J]. Bioinformatics, 19(12): 1572-1574.
Sahlin K, Chikhi R, Arvestad L. 2016. Assembly scaffolding with PE-contaminated mate-pair libraries[J]. Bioinformatics, 32(13): btw064.Sahlin K, Vezzi F, Nystedt B,etal. 2014. BESST-efficient scaffolding of large fragmented assemblies[J]. BMC Bioinformatics, 15(1): 281.
Sánchezgracia A, Maside X, Charlesworth B. 2005.High rate of horizontal transfer of transposable elements inDrosophila[J]. Trends in Genetics, 21(4): 200.
Sinkins S. 2007. Genome sequence ofAedesaegypti, a major arbovirus vector[J]. Science, 316(5832): 1718-1723.
Smit A, Hubley R. 2008-2015. RepeatModeler open-1.0[EB/OL]. URL: http: //www. repeatmasker. org.
Smit A, Hubley R, Green P. 2013-2015. RepeatMasker open-4.0[EB/OL]. URL: http://www. repeatmasker. org.
Sormacheva I, Smyshlyaev G, Mayorov V,etal. 2012. Vertical evolution and horizontal transfer of CR1 non-LTR retrotransposons and Tc1/mariner DNA transposons in Lepidoptera species[J]. Molecular Biology & Evolution, 29(12): 3685-3702.
Sun C, Mueller RL. 2014. Hellbender genome sequences shed light on genomic expansion at the base of crown salamanders[J]. Genome Biology & Evolution, 6(7): 1818-1829.
Tamura K, Peterson D, Peterson N,etal. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J]. Molecular Biology & Evolution, 28(10): 2731-2739.
Tay WT, Behere GT, Batterham P,etal. 2010. Generation of microsatellite repeat families by RTE retrotransposons in lepidopteran genomes[J]. BMC Evolutionary Biology, 10(1): 144.
Terrapon N, Li C, Robertson HM,etal. 2014. Molecular traces of alternative social organization in a termite genome[J]. Nature Communications, 5(6183): 3636.
Thompson JD. 1999.CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice[J]. Nucleic Acids Research, 22(22): 4673-4680.
Webb CHT, Riccitelli NJ, Ruminski DJ,etal. 2011. Widespread occurrence of self-cleaving ribozymes[J]. Science, 326(5955): 953.
Zhang HH, Feschotte C, Han MJ,etal.2014. Recurrent horizontal transfers of Chapaev transposons in diverse invertebrate and vertebrate animals[J]. Genome Biology & Evolution, 6(6): 1375.
Zhao FQ, Qi J, Schuster SC. 2009. Tracking the past: interspersed repeats in an extinct Afrotherian mammal,Mammuthusprimigenius[J]. Genome Research, 19(19): 1384-1392.
AnalysisofRepetitiveSequencesinPeriplanetaamericananaGenome
MOU Biqin1, YAN Chaochao1, LI Wujiao1, LI Jing1, SHEN Yongmei2, YUE Bisong1*
(1. Key Laboratory of Bio-resources and Eco-environment, Ministry of Education, College of Life Sciences, Sichuan University, Chengdu 610064, China; 2. Sichuan Key Laboratory of MedicinalPeriplanetaamericana, Chengdu 610081, China)
2017-02-22接受日期2017-05-09
牟必琴, 女, 硕士研究生, 研究方向:基因和遗传信息分析, E-mail:435077636@qq.com
*通信作者Corresponding author, E-mail:bsyue@scu.edu.cn
10.11984/j.issn.1000-7083.20170051
Q78
A
1000-7083(2017)05-0540-08
猜你喜欢
林业科学(2022年1期)2022-03-23
浙江农林大学学报(2021年3期)2021-07-12
波谱学杂志(2021年1期)2021-03-12
科学(2020年3期)2020-11-26
中国农业科学(2020年7期)2020-04-11
文萃报·周二版(2018年22期)2018-09-18
中学生数理化·高一版(2018年6期)2018-07-09
故事作文·高年级(2017年11期)2017-11-15
武昌理工学院学报(2017年1期)2017-05-11
浙江农林大学学报(2016年6期)2016-12-12
