
肾脏脂肪囊多形性脂肪肉瘤1例报告并文献复习
2017-10-10 01:00:42张帆王春杨陈光富郭刚肖克峰张旭
微创泌尿外科杂志 2017年5期
张帆 王春杨 陈光富 郭刚 肖克峰 张旭
1中国人民解放军总医院泌尿外科 100853 北京2深圳市人民医院泌尿外科
肾脏脂肪囊多形性脂肪肉瘤1例报告并文献复习
张帆1王春杨1陈光富1郭刚1肖克峰2张旭1
1中国人民解放军总医院泌尿外科 100853 北京2深圳市人民医院泌尿外科
目的探讨肾脏脂肪囊多形性脂肪肉瘤的的临床特点、病理特点、诊治方法及预后。方法报告1例肾脏脂肪囊多形性脂肪肉瘤患者的临床资料,结合相关文献复习并讨论。结果患者,男,67岁。因影像学发现右肾区占位入院,接受腹腔镜下右肾根治性切除术,病理诊断为肾脏脂肪囊多形性脂肪肉瘤,免疫组化染色显示S-100(弱+),MDM2(+),CDK4(+),CD10(+),muscle(-),CK(-)。术后随访17个月无原位复发及远处转移。结论肾脏脂肪囊多形性脂肪肉瘤临床罕见,具有高度恶性和侵袭性,易复发及转移,预后较差。确诊需依靠影像学及病理免疫组化检查,手术完整切除肿瘤是目前有效的治疗方法。
肾脏肿瘤;多型性脂肪肉瘤;肾切除术
脂肪肉瘤(liposarcoma, LS)是起源于软组织的少见恶性肿瘤类型,而多形性脂肪肉瘤(pleomorphic liposarcoma, PLS)更是其中少见的病理亚型,占所有LS的5%以下[1]。PLS好发于腹膜后间隙器官周围,发生于肾脏脂肪囊的目前报道极少。我科2013年收治1例肾脏脂肪囊多形性脂肪肉瘤(pleomorphic liposarcoma of renal adipose capsule, PLSRAC)患者,现报告如下,并结合相关文献进行复习。
1 病例资料

图1 MRI检查提示肿瘤位于肾脏脂肪囊内(箭头所指为病灶)
患者,男,67岁。因体检时发现右肾区占位性病变1周于2013年5月23日入我科治疗。患者2013年5月17日体检时,MRI提示右肾区占位性病变(图1),病变位于右肾中下部背侧包膜下,横轴位大小约2.7 cm,累及相邻肾后筋膜,呈略长T2、略长T1不均匀信号,弥散加权(diffusion-weighted, DWI)为高信号,动脉期呈轻度强化,以后各期呈逐渐显著的略不均匀强化。病程中患者无任何不适症状,饮食睡眠无异常,体重无明显变化。入院后查体:腹软,全腹无压痛反跳痛,腹部未触及肿块,双肾均未触及;实验室检查:肿瘤标记物未见异常指标。血尿便常规、血生化、凝血常规均未见异常指标;ECT肾图:GFR(左肾28.1 ml/min,右肾33.4 ml/min);PET/CT提示:右肾高代谢结节,SUVmax10.97,延迟40 min后,放射性摄取有所增强,SUVmax12.65,考虑原发恶性肿瘤性病变。为进一步明确诊断行超声引导下右肾区占位性病变穿刺活检术,穿刺病理结果:肉瘤样癌可能性大,但不能除外其他非上皮源性恶性肿瘤。免疫组化示:vimentin(+),CD10(+),CK(少数+),CK7(-),CK18(-),AE1(-),EMA(-),CD68(-),CD34(-),CD117(-),HMB45(-),SMA(-),muscle(-),CD20(-),CD30(-)。经积极术前准备于2013年6月12日在全麻下行腹腔镜下右肾根治性切除术,术后患者恢复良好。切除组织行常规苏木素-伊红染色及免疫组化检查。
2 病理结果
2.1肉眼所见
手术切除右侧肾脏、部分输尿管及部分粘连之筋膜,大小13 cm×9 cm×5 cm。肾脏大小10.0 cm×5.0 cm×4.5 cm,脂肪囊不易剥离。其余肾皮质、髓质分界清楚,皮质厚0.6 cm,肾盂黏膜灰白、光滑,输尿管长5 cm,直径0.4 cm。肾门处未见淋巴结。肿瘤大小4.5 cm×3.0 cm×2.8 cm,切面灰白、质硬。肿瘤挤压肾皮质,在脂肪组织中浸润性生长,界限不清。肾皮质内瘤体部分呈结节状,范围为1.5 cm×1.0 cm(图2)。
2.2HE染色
高倍镜下肿瘤细胞呈弥漫分布,形状不规则,可见多核巨细胞散在分布,核大深染,可见病理性核分裂相。瘤周肾实质淤血、灶状出血,被膜下较多炎细胞浸润(图3A)。
2.3免疫组化染色
免疫组化染色显示肿瘤细胞S-100(弱+),MDM2(+),CDK4(+),CD10(+),muscle(-),CK(-)(图3B、C、D)。

肉眼见肿瘤位于肾脏脂肪囊内,呈浸润性生长,挤压肾皮质(红圈内为肿瘤)。
图2病理标本
2.4随访资料
明确诊断后患者接受外放疗,方案:TomoTherapy,PTV1=60 Gy,PTV2=50 Gy,共25次。手术后随访17个月,患者无原位复发及远处转移(图4)。
3 讨论
3.1概述
软组织肉瘤(soft tissue sarcoma, STS)是起源于间叶组织的少见肿瘤,约占全部成人恶性肿瘤的1%[2]。LS占所有STS的17%~25%[3]。根据WHO 2013版软组织肿瘤组织学分类,LS可细分为去分化脂肪肉瘤(dedifferentiated liposarcoma, DLS)、黏液样脂肪肉瘤(myxoid liposarcoma, MLS)、多形性脂肪肉瘤(pleomorphic liposarcoma, PLS)、非典型脂肪瘤/高分化脂肪肉瘤(atypical lipomatous tumor/well differentiated liposarcoma, ALT/WDLS)四种亚型。其中PLS约占所有LS的5%[4]。有报道PLS可发生于2~98岁任何年龄[5],但主要以50岁以上成人为主,中位发病年龄在53~63岁[6, 7],男女比例约为1.26∶1[8]。本例为67岁男性患者。
3.2临床特点
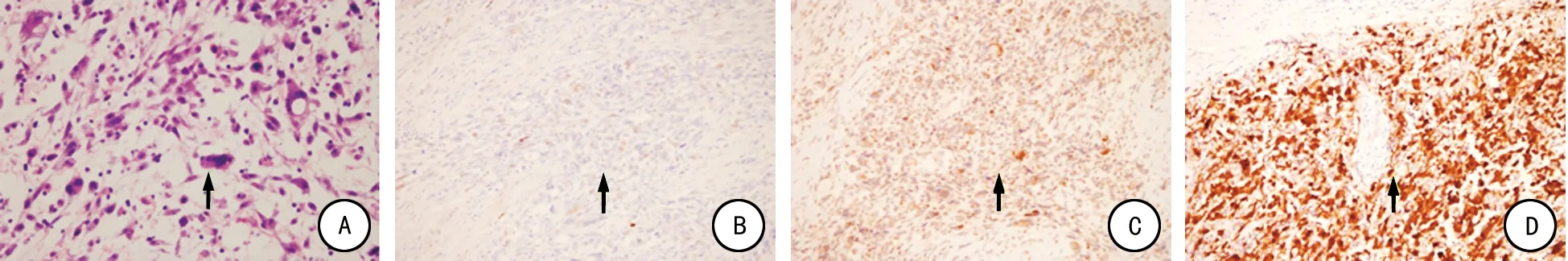
A:HE×400;B:S-100(弱+);C:MDM2(+);D:CDK4(+)。
图3HE染色及免疫组化染色
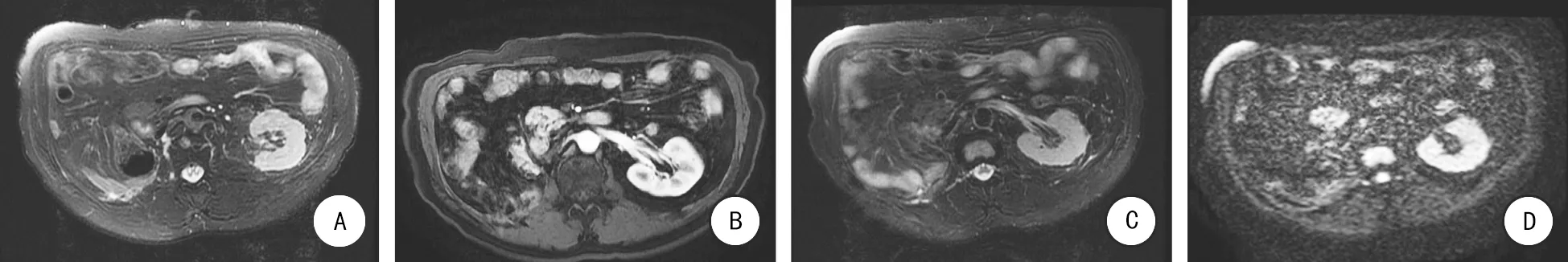
A:术后3个月T2WI;B:术后3个月增强扫描;C:术后6个月T2WI;D:术后6个月DWI。
图4术后MRI复查
PLS好发生于深部软组织间隙[6],其中发生于下肢和上肢较常见,发生率分别约为47%和18%[4]。另外有7%~10%发生于腹膜后[5],主要见于肾脏、脾肾之间、胰腺、肾上腺、十二指肠等器官[9]。也有少量报道见于纵隔[10]、心包[11, 12]、肺[13]、乳腺[14]、骨[15, 16]、皮下组织[17]等。PLSRAC起源于肾周脂肪组织,沿腹膜后间隙发展,一般不侵犯集合系统,血尿少见。患者早期无特异临床症状及体征,肿瘤在初期时生长缓慢甚至出现“惰性生长”,临床很难通过体检发现,有时在影像学检查时偶然发现。但肿瘤经过一段迟缓生长期后,其生长速度迅速加快,呈现“激怒”式生长[18]。由于肿瘤位于深部组织间隙或者腹膜后间隙,生长空间较大,很多患者往往是在偶然发现包块、出现局部疼痛、肿胀或者出现运动系统、泌尿系统、呼吸系统、消化系统、神经系统等压迫症状时才就诊。也有患者以体重减轻、食欲下降、低热、贫血等全身症状就诊。有报道约有94%的PLS直径超过5 cm,60%的PLS直径超过10 cm,20%~40%的PLS直径超过20 cm[5, 19, 20],而小于5 cm的PLS较少见[9]。本例患者就诊时无明显主观症状,查体时也无任何阳性体征,仅在体检时影像学发现肿瘤,肿瘤直径小于5 cm。
3.3诊断及鉴别诊断
由于PLS患者早期缺乏典型症状体征,因此诊断主要依赖影像学检查如B超、CT、MRI、PET/CT等。B超由于其无创特点可以作为初筛手段,特别是在PLS增大并出现腹部症状后的鉴别诊断中,超声下PLS多为低回声实性占位,内回声不均匀,CDFI显示肿瘤内部及周边可见点状血流信号[21, 22]。但由于超声检查缺乏特异性,不推荐作为主要确诊手段。CT对诊断PLS有重要意义,可显示位置、大小、浸润范围、与周围组织关系,并且可显示肿瘤是否有液化、坏死、囊性变及钙化等改变。在CT图像上,PLS表现为以实性为主混合含量不等的脂肪成分肿块,中间可见条索状分隔,静脉注射对比剂后肿瘤实性部分呈显著不均匀强化[23, 24]。有报道可以根据CT影像上肿瘤所含脂肪量的不同将PLS与LS其他亚型相鉴别:WDLS在CT上具有较丰富的脂肪成分;DLS几乎没有脂肪成分,取而代之的是一些局灶性结节以及软组织密度成分[18, 25];由于PLS恶性程度高,并且报道易转移至肺部,因此行腹部CT的同时进行肺部CT扫描,以排除肺部是否有转移病灶是必要的[2]。MRI检查可以显示肿瘤与周围结构关系,与CT相比,MRI包含更多信息量。在MRI影像上,PLS表现为边界清楚的肿瘤,内部信号不均匀,T1WI为低信号,T2WI为高信号,有时肿瘤内部可出现坏死区域,在T2WI上呈现为高信号,增强后肿瘤出现不均匀明显强化[26, 27]。PET/CT兼有PET和CT两种影像学技术的优点,同时可对全身各组织器官进行检查,有助于发现远处转移病灶。国内丁重阳等[28]报道在PET/CT上,分化好的脂肪肉瘤CT值在-20 HU以下且有FDG摄取不均匀性增高。国外Tuan等[26]报道PET/CT联合FDG可将高级别的脂肪肉瘤与正常软组织进行区分,但是对于区分中低级别的脂肪肉瘤和正常软组织较为困难。肿瘤穿刺活检可用于影像学诊断有疑议的患者,通常在CT或者超声引导下进行,Stattaus等[29]报道共轴穿刺活检可以鉴别95.9%的恶性病变。但鉴于穿刺活检具有针道种植转移的风险以及非100%的诊断准确率,因此穿刺活检仅推荐用于为选择治疗方式而需要确定肿瘤的确切组织学类型,或者对影像学的诊断存在争议时使用[25]。本例患者MRI提示肿瘤呈高T2、略高T1信号,且信号不均匀,增强后肿瘤呈不均匀强化。
PLSRAC需要与以下疾病进行鉴别,①肾细胞癌:肾细胞癌是肾脏最常见恶性肿瘤类型,起源于肾小管上皮细胞,CT及MRI扫描可见肿瘤呈圆形或类圆形,有假包膜,增强扫描后肿瘤明显强化;②肾血管平滑肌脂肪瘤:又称错构瘤,CT平扫可见肿瘤内部含有低密度脂肪成分,MRI脂肪抑制相位可以更准确鉴别肿瘤内部的脂肪信号与实性成分信号。此外,肾血管平滑肌脂肪瘤的供血血管扩张且多为单支供血,CT和MRI上表现为与正常肾实质相连的桥接血管, CTA和MRA有助于肿瘤内扩张血管的显示。③恶性纤维组织细胞瘤:PLS与本病有时鉴别困难。PLS常可见脂肪母细胞分化,其内部有边界清楚且透明的空泡结构,免疫组化S-100呈阳性。恶性纤维组织细胞瘤有时表现为多形性,伴有瘤巨细胞,内部也可存在空泡结构,但边界不清,空泡内有半透明的黏多糖物质,免疫组化染色S-100呈阴性[30]。
3.4病理特点
大体观肿瘤呈分叶状或多结节状,质硬,边界不清,切面呈白色、黄色或黄白色相间,内部偶可见黏液改变以及坏死[7]。光镜下可见肿瘤与周围组织界限不清,有大小不等的脂源性或非脂源性区域,两者没有固定比例。脂源性区域由恶性或者多形性脂肪细胞以及多空泡脂肪母细胞构成,核异型明显,可见瘤巨细胞。非脂源性区域在不同肿瘤间差异较大,有的呈现多形恶性纤维组织细胞瘤样改变,有的为中至高级别黏液纤维肉瘤样改变,有的为横纹肌肉瘤样并可见核深染的多核巨细胞。几乎所有病例都能看到背景中有多空泡脂肪母细胞存在[7]。免疫组化染色显示在40%~50%病例中可以观察到SMA局灶阳性,13%~19%病例中desmin局灶阳性,约40%病例中CD34阳性,约26%的病例中EMA阳性,约13%病例中CD68阳性,但以上均不特异[6, 7]。SMA阳性可以在一些病例的脂肪母细胞中观察到,但比例差异较大。而S-100局灶阳性可以在大多数病例中观察到[4]。因此本病需要结合影像学及病理学免疫组化加以确诊。本例患者免疫组化显示S-100(弱+),MDM2(+),CDK4(+),CD10(+),muscle(-),CK(-),在穿刺活检组织的免疫组化染色中SMA显示为阴性。
3.5治疗
目前针对PLSRAC的报道极少,已报道的病例中治疗方式以手术完整切除为主,但限于肿瘤未出现远处转移的患者[18, 31]。手术在切除原发病灶的同时完整切除临近受累组织结构,包括腹膜、腰大肌、锥旁肌肉、腹壁等。腹腔镜和开放手术均是推荐的手术方式,但需要结合患者肿瘤大小以及身体情况等综合选择[32]。由于PLS是放射线敏感性肿瘤,也有一些研究认为术后对术区进行辅助性放疗可以降低局部复发风险并延长无复发间期[33, 34],但术后局部放疗并未使患者在总生存期方面获益[35, 36],另外Fein等[37]报道术后对术区的放疗会增加对胃肠道的副作用。对于广泛转移无法切除的PLS,全身化疗是目前可选择的治疗手段;对于局限性的PLS患者,全身化疗或新辅助化疗的治疗效果目前仍存在争议。有学者报道,化疗/新辅助化疗不能使PLS患者受益[38];Donahue等[39]研究发现,组织学高级别的PLS接受术后化疗可以显著改善5年疾病特异性生存率。此外,也有化疗联合局部放疗的报道[40, 41],可行性和安全性均比较乐观。本例患者采用腹腔镜下右肾根治性切除术,术后给予术区辅助性外放疗,术后随访17个月无局部复发及远处转移,但远期预后情况尚需要进行后续长期的随访。
3.6预后
目前报道的PLS患者5年生存率为20%~63%[4, 6, 7, 42, 43]。在PLS患者中,肿瘤局部复发较远处转移更为常见[18]。已报道的PLS局部复发率为25%~48%,并且原发于腹部的PLS较四肢的PLS更容易术后复发[6, 7, 42],可能的原因是四肢的肿瘤较腹部的肿瘤在手术切除时更容易达到切缘阴性标准,而切缘阴性与否是影响PLS患者术后生存时间和是否复发的重要因素[19]。有报道PLS转移率为32%~50%,常见的部位是肺、骨、肝、皮肤[6, 7, 42]。另外,Gebhard等[6]、Hornick等[7]以及Wang等[4]分别通过多因素分析发现,PLS患者预后差的主要影响因素是年龄大于60岁、肿瘤直径超过10 cm、肿瘤位于深部组织、手术切缘阳性、肿瘤坏死、术后局部复发和远处转移。
综上所述,PLS是一种高度侵袭、高度恶性的实体肿瘤,并且具有早期转移和复发的特点,预后大多较差[6, 43]。PLSRAC罕见,该病早期缺乏典型症状体征,影像学及病理学免疫组化检查可作为主要确诊手段。对于本病应做到早期发现和早期诊断,在患者身体情况允许情况下,积极尽早手术完整切除肿瘤,争取提高患者预后。
[1] Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol, 2012,40(12):1122-1131.
[2] Dei Tos AP. Liposarcomas: diagnostic pitfalls and new insights. Histopathology, 2014,64(1):38-52.
[3] Kollar A, Benson C. Current management options for liposarcoma and challenges for the future. Expert Rev Anticancer Ther, 1994,14(3):297-306.
[4] Wang L, Ren W, Zhou X, et al. Pleomorphic liposarcoma: a clinicopathological, immunohistochemical and molecular cytogenetic study of 32 additional cases. Pathol Int, 2013,63(11):523-531.
[5] Liles JS, Tzeng CW, Short JJ, et al. Retroperitoneal and intra-abdominal sarcoma. Curr Probl Surg, 2009,46(6):445-503.
[6] Gebhard S, Coindre JM, Michels JJ, et al. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: a study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol, 2002,26(5):601-616.
[7] Hornick JL, Bosenberg MW, Mentzel T, et al. Pleomorphic liposarcoma-Clinicopathologic analysis of 57 cases. Am J Surg Pathol, 2004,28(10):1257-1267.
[8] Lehnert T, Cardona S, Hinz U, et al. Primary and locally recurrent retroperitoneal soft-tissue sarcoma: Local control and survival. EJSO, 2009,35(9):986-993.
[9] Ferrario T, Karakousis CP. Retroperitoneal sarcomas: grade and survival. Arch Surg, 2003,138(3):248-251.
[10] Hahn HP, Fletcher CD. Primary mediastinal liposarcoma: clinicopathologic analysis of 24 cases. Am J Surg Pathol, 1994,31(12):51-52.
[11] Wang JG, Wei ZM, Liu H, et al. Primary pleomorphic liposarcoma of pericardium. Interact Cardiovasc Thorac Surg, 2010,11(3):325-327.
[12] Yie K, Ryu SM, Cho SJ. Metastasis after resection of huge primary pericardial pleomorphic liposarcoma with cardiopulmonary bypass. Thorac Cardiovasc Surg, 2010,58(2):113-115.
[13] Ibe T, Otani Y, Shimizu K, et al. Pulmonary pleomorphic liposarcoma. Jpn J Thorac Cardiovasc Surg, 2005,53(8):443-447.
[14] Mardi K, Gupta N. Primary pleomorphic liposarcoma of breast:a rare case report. Indian J Pathol Microbiol, 1994,54(1):51-52.
[15] Torigoe T, Matsumoto T, Terakado A, et al. Primary pleomorphic liposarcoma of bone: MRI findings and review of the literature. Skeletal Radiol, 2006,35(7):536-538.
[16] Rasalkar DD, Chow LT, Chu WC, et al. Primary pleomorphic liposarcoma of bone in an adolescent: imaging features of a rare entity. Pediatr Radiol, 2011,41(10):1342-1345.
[17] Gardner JM, Dandekar M, Thomas D, et al. Cutaneous and subcutaneous pleomorphic liposarcoma: a clinicopathologic study of 29 cases with evaluation of MDM2 gene amplification in 26. Am J Surg Pathol, 2012,36(7):1047-1051.
[18] Vijay A, Ram L. Retroperitoneal liposarcoma: a comprehensive review. Am J Clin Oncol, 1994,38(2):213-219.
[19] Lewis JJ, Leung D, Woodruff JM, et al. Retroperitoneal soft-tissue sarcoma - Analysis of 500 patients treated and followed at a single institution. Ann Surg, 1998,228(3):355-363.
[20] Bonvalot S, Rivoire M, Castaing M, et al. Primary retroperitoneal sarcomas: a multivariate analysis of surgical factors associated with local control. J Clin Oncol, 2009,27(1):31-37.
[21] 王蕾,宋光.原发性腹膜后脂肪肉瘤的超声和CT诊断.中国疗养医学,2013,22(12):1068-1069.
[22] Ishida H, Naganuma H, Konno K, et al. Retroperitoneal liposarcoma: sonographic findings. Abdom Imaging, 2000,25(5):554-558.
[23] 江心,张杰,林洁,等.原发性腹膜后脂肪肉瘤CT表现与病理对照分析.放射学实践,2013,28(9):964-967.
[24] 杨志勇,高兵.原发性腹膜后脂肪肉瘤的CT诊断.中国医药导刊,2014,16(6):1080-1081.
[25] Olimpiadi Y, Song S, Hu JS, et al. Contemporary management of retroperitoneal soft tissue sarcomas. Curr Oncol Rep, 2015,17(8):39.
[26] Tuan J, Vitolo V, Vischioni B, et al. Radiation therapy for retroperitoneal sarcoma. Radiol Med, 2014,119(10):790-802.
[27] 林翠君,李丽红,黄春榆,等.脂肪肉瘤的CT、MRI表现与病理学对照.中国CT和MRI杂志,2015,13(8):108-111.
[28] 丁重阳,李天女,吉爱兵.原发性腹膜后恶性肿瘤的18F-FDG PET/CT显像特征.中国临床医学影像杂志,2014,25(9):674-676.
[29] Stattaus J, Kalkmann J, Kuehl H, et al. Diagnostic yield of computed tomography-guided coaxial core biopsy of undetermined masses in the free retroperitoneal space: single-center experience. Cardiovasc Intervent Radiol, 2008,31(5):919-925.
[30] 唐涛,范嫏娣,林建韶.多形性脂肪肉瘤的临床病理特征.诊断病理学杂志,2006,13(6):457-459.
[31] 孔庆龙,李荣,郑伟,等.肾周脂肪肉瘤的诊断和外科治疗.中国实用外科杂志,2007,27(4):284-285.
[32] Shalhav AL, Chan SW, Bercowsky E, et al. Laparoscopic exploration in the management of retroperitoneal masses. JSLS, 1999,3(3):209-214.
[33] Lahat G, Tuvin D, Wei C, et al. New perspectives for staging and prognosis in soft tissue sarcoma. Ann Surg Oncol, 2008,15(10):2739-2748.
[34] Le Péchoux C, Musat E, Baey C, et al. Should adjuvant radiotherapy be administered in addition to front-line aggressive surgery (FAS) in patients with primary retroperitoneal sarcoma? Ann Oncol, 2013,24(3):832-837.
[35] Tseng WH, Martinez SR, Do L, et al. Lack of survival benefit following adjuvant radiation in patients with retroperitoneal sarcoma: a SEER analysis. J Surg Res, 2011,168(2):e173-e180.
[36] Choi AH, Barnholtz-Sloan JS, Kim JA. Effect of radiation therapy on survival in surgically resected retroperitoneal sarcoma: a propensity score-adjusted SEER analysis. Ann Oncol,2012,23(9):2449-2457.
[37] Fein DA, Corn BW, Lanciano RM, et al. Management of retroperitoneal sarcomas: does dose escalation impact on locoregional control? Int J Radiat Oncol Biol Phys, 1995,31(1):129-134.
[38] Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol, 2007,8(7):595-602.
[39] Donahue TR, Kattan MW, Nelson SD, et al. Evaluation of neoadjuvant therapy and histopathologic response in primary, high-grade retroperitoneal sarcomas using the sarcoma nomogram. Cancer, 2010,116(16):3883-3891.
[40] Pisters PW, Ballo MT, Fenstermacher MJ, et al. Phase I trial of preoperative concurrent doxorubicin and radiation therapy, surgical resection, and intraoperative electron-beam radiation therapy for patients with localized retroperitoneal sarcoma. J Clin Oncol, 2003,21(16):3092-3097.
[41] Petrioli R, Coratti A, Correale P, et al. Adjuvant epirubicin with or without Ifosfamide for adult soft-tissue sarcoma. Am J Clin Oncol, 2002,25(5):468-473.
[42] Downes KA, Goldblum JR, Montgomery EA, et al. Pleomorphic liposarcoma: a clinicopathologic analysis of 19 cases. Mod Pathol, 2001,14(3):179-184.
[43] Dodd LG, Sara Jiang X, Rao K, et al. Pleomorphic liposarcoma: a cytologic study of five cases. Diagn Cytopathol, 2015,43(2):138-143.
Pleomorphicliposarcomaofrenaladiposecapsuleacasereportandliteraturesreview
ZhangFan1WangChunyang1ChenGuangfu1GuoGang1XiaoKefeng2ZhangXu1
(1Department of Urology, Chinese PLA General Hospital, Beijing 100853, China;2Department of Urology, Shenzhen People's Hospital)
Zhang Xu, xzhang@foxmail.com
Objective: To investigate the clinical characteristics, pathological features, diagnosis, treatment, and prognosis of pleomorphic liposarcoma of renal adipose capsule.MethodsThe clinical data of a patient with pleomorphic liposarcoma of renal adipose capsule were reported, and related literatures were reviewed.ResultsA 67-year-old male patient was diagnosed as having renal neoplasm by imageological examination. The patient underwent laparoscopic radical nephrectomy on right kidney. Pathological diagnosis showed the pleomorphic liposarcoma of renal adipose capsule. Immunohistochemical stain showed positive results for S-100, MDM2, CDK4 and CD10, while negative for muscle and CK. The patient was followed-up for 17 months, and no recurrence or metastasis was observed.ConclusionsPleomorphic liposarcoma of renal adipose capsule is rarely seen clinically. It is an aggressive, high grade neoplasm with a tendency for recurrence and metastases and often carries a poor prognosis. The definite diagnosis mainly depends on imageological examination and immunohistochemical stain. A complete resection surgery is the best treatment method.
renal neoplasm; pleomorphic liposarcoma; nephrectomy
R737.11
A
张旭,xzhang@foxmail.com
2016-11-10
10.19558/j.cnki.10-1020/r.2017.05.011
猜你喜欢
中国医学影像技术(2022年12期)2022-12-28 09:46:48
中国临床医学影像杂志(2022年2期)2022-05-25 13:24:54
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
蚌埠医学院学报(2019年9期)2019-10-14 07:00:44
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:06
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:04
浙江医学(2018年16期)2018-09-08 05:58:00
解剖学杂志(2018年2期)2018-05-21 10:01:13
中华老年口腔医学杂志(2016年3期)2017-01-15 14:24:59
