
SO42-/ZrO2-SiO2固体超强酸制备工艺研究
2017-09-28 09:34:52徐凤华翟祝贺田孟超李爱梅赵亚楠
河南科技学院学报(自然科学版) 2017年4期
徐凤华,翟祝贺,田孟超,李爱梅,赵亚楠
(中原工学院,河南郑州450007)
SO42-/ZrO2-SiO2固体超强酸制备工艺研究
徐凤华,翟祝贺,田孟超,李爱梅,赵亚楠
(中原工学院,河南郑州450007)
采用浸渍法制备SO42-/ZrO2-SiO2固体超强酸,并以廉价的1,4-丁二醇和工业废气在吸收过程中产生的溴化氢为原料,以合成的SO42-/ZrO2-SiO2固体超强酸为催化剂合成用途广泛的1,4-二溴丁烷.采用正交试验研究了SO42-/ZrO2-SiO2固体超强酸制备过程中的复配比、焙烧温度、焙烧时间、硫酸浓度等因素对其催化性能的影响,以1,4-二溴丁烷的收率为考核指标,确定了SO42-/ZrO2-SiO2固体超强酸的最佳制备工艺.结果表明:当n(Zr)∶n(Si)为1∶4、陈化时间12 h、预焙烧温度200℃、焙烧温度550℃、焙烧时间3 h、H2SO4浓度1.00 mol/L、m(前驱体)∶V(H2SO4)=1∶10时制备的固体超强酸的催化性能最佳.
固体超强酸;SO42-/ZrO2-SiO2;1,4-丁二醇;1,4-二溴丁烷
Abstract:In this article,SO42-/ZrO2-SiO2solid superacid were synthesized by impregnation method,and the wildly used 1,4-dibromobutane was synthesized with the prepared solid superacidas catalystutilizing raw material of inexpensive 1,4-butyl glycol and industrial abandoned hydrogen bromide produced during absorption process. Orthogonal test was used to study the effects of compound ratio,calcination temperature,calcination time and sulfuric acid concentration on the catalytic performance of SO42-/ZrO2-SiO2solid superacid.The results indicated that:When n(Zr)∶n(Si)is 1∶4,aging time is 12 hour,pre-calcination temperature is 200℃,roasting temperature is 550℃, roasting time 3 hour,H2SO4concentration is 1.00 mol/L,m(precursor)∶V(H2SO4)=1∶10,the solid superacid prepared above performs the best in catalytic performance.
Key words:solid superacid;SO42-/ZrO2-SiO2;1,4-butyl glycol;1,4-dibromobutane
酸作为一种催化剂被广泛应用于许多重要的化学反应中.在酸作为催化剂的反应中反应速度一般都与酸强度成正比,因此如何提高酸强度并加以利用成为现代化学研究的热点之一.超强酸即酸度超过100%硫酸的酸度,用Hammett指示剂的酸度函数H0表示,100%硫酸的H0=-11.93,对于H0<-11.93的酸均为超强酸,H0越小,该超强酸的酸强度越强[1].超强酸按形态可分为液体和固体两种.液体超强酸大多是由氟化合物(氟化氢、氟磺酸等)按照一定比例混合复配而成的;固体超强酸可以分为含卤素和不含卤素两大类,前者是由氟氧化物负载在多孔的无机物表面构成的,后者则是将硫酸根吸附在金属或非金属氧化物表面上而形成的.
相比较于另外两类超强酸,硫酸型固体超强酸具有多个优点,如:制备工艺简单;催化剂不含卤素离子,对环境污染小;催化剂种类多,酸强度高;适用性广;易回收再利用等.所以固体超强酸的制备成为研究的热点.本文以ZrOCl2和Na2SiO3为原料,通过沉淀浸渍法制备SO42-/ZrO2-SiO2固体超强酸,并以此为催化剂,合成用途广泛的1,4-二溴丁烷,以1,4-二溴丁烷的收率为考核指标探究SO42-/ZrO2-SiO2固体超强酸制备的最佳工艺.
1 材料与方法
1.1 药品及仪器
红外吸收光谱使用Tensor-37红外光谱仪(德国Bruker光谱仪器公司)测定;固体超强酸的比表面积使用ASIQM000000-6比表面积测定仪(Quantachrome)测定;1,4-二溴丁烷的纯度用GC8100Ⅱ气相色谱仪(滕州市经纬分析仪器有限责任公司)和2W双目阿贝折光仪(上海光学五厂)测定.
二氯氧化锆(河南佰利联化学股份有限公司);1,4-丁二醇(河南义马煤业集团开祥化工有限公司);硅酸钠;无水碳酸钠,硫酸,溴化氢,氨水均购自国药集团上海化学试剂有限公司(分析纯),试剂未经处理.
1.2 反应机理
1.2.1 固体超强酸的酸性中心模型某些金属氧化物本身是没有酸性的,只有经过含有SO42-离子的溶液浸渍后,使得金属氧化物表面上的硫为最高价态时,催化剂才能表现出超强酸性,这是SO42-/MxOy型固体超强酸中心形成的必要条件[2].通常情况下,金属氧化物与SO42-有单配位、螯合式双配位、桥式双配位三种配位形态[3].文献[4]表明,在SO42-/MxOy型固体超强酸中,SO42-与金属氧化物之间以后两种配位作用为主.催化剂超强酸的酸性中心是由于金属氧化物与SO42-发生强的诱导效应产生的金属离子[5],在焙烧过程中离子型的S=O键转变为共价键形式的S=O键,S=O基团为强的吸电子基团,由于共价键的吸电子诱导效应,使得M-O键上的电子云密度降低,从而产生强的L型(路易斯酸)酸性中心,进而表现出超强酸性,水分子或者OH-在L型酸性中心发生吸附解离而产生B型(布氏酸)酸性中心.B型和L型酸性中心的协同作用使催化剂的酸性高于100%硫酸的酸性.
1.2.2 固体超强酸催化合成1,4-二溴丁烷的反应机理大部分伯醇与氢卤酸的反应是按照SN2机理进行的[6],反应历程如下:

1.3 试验步骤
1.3.1 SO42-/ZrO2-SiO2固体超强酸前驱体的制备按照n(Zr)与n(Si)的比例称取一定量的ZrOCl2· 8H2O和Na2SiO4·9H2O,分别用蒸馏水溶解,再分别用氨水和1 mol/L硫酸溶液做沉淀剂,调节两种溶液的pH值,用pH试纸测得两种溶液的pH值为7~8,将这两种沉淀的母液充分搅拌混合均匀,在室温条件下陈化24 h,过滤,用蒸馏水充分洗涤,直至用AgNO3溶液检测洗涤液中无Cl-为止,得到固体超强酸的前驱体. 1.3.2 SO42-/ZrO2-SiO2固体超强酸制备将所得的前驱体在110℃下烘干,充分研磨,过150目筛网,在200℃下预焙烧2 h,按比例加入一定量的硫酸溶液搅拌2 h后,静置2 h,用砂芯漏斗抽滤,100℃下烘干,在一定温度下焙烧数小时,即制得SO42-/ZrO2-SiO2固体超强酸.
1.3.3 1,4-二溴丁烷的合成在100 mL三口烧瓶中加入2 g固体超强酸、9 g1,4-丁二醇,升温至90℃,将67 g质量分数为40%的氢溴酸通过滴液漏斗缓慢滴加到配备有球型回流冷凝管、温度计、电磁加热搅拌器的100 mL三口烧瓶内,110℃下搅拌回流反应6 h.反应完毕后将回流装置改为蒸馏装置,收集140℃以下的馏分.将收集的馏分静置分层,取下层油状物,用质量分数为5%的碳酸氢钠溶液充分洗涤至中性,接着用蒸馏水洗涤.将洗涤后的产物在压力为0.09 MPa、温度85℃左右条件下旋转蒸发2 h.将目标产物进行减压蒸馏,称质量,计算收率并进行结构表征.
2 结果与分析
2.1 陈化时间对固体超强酸催化性能的影响
在n(Zr)∶n(Si)=1∶3,预焙烧温度200℃,焙烧温度550℃,焙烧时间3 h,硫酸浓度1 mol/L的条件下,改变陈化时间,制备固体超强酸,探究陈化时间对固体超强酸催化性能的影响.结果见表1.
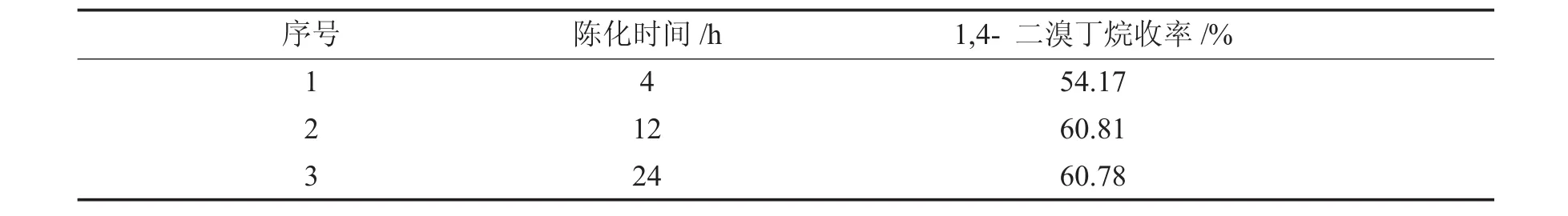
表1 陈化时间对固体超强酸催化性能的影响Tab.1 Effect ofagingtime on catalytic performance ofsolid superacid
陈化是为了在陈化过程中除去沉淀中包含的杂质,让晶体生长增大,并且使晶体的粒径均匀分布.由试验结果可以看到,当陈化时间为12 h和24 h时,1,4-二溴丁烷的收率几乎相同,但高于陈化时间为4 h的收率.考虑生产效率因素,选择最佳陈化时间为12 h.
2.2 预焙烧对固体超强酸催化性能的影响
由于浸渍的时候H2SO4能与固体超强酸的前驱体发生反应,使得部分前驱体溶解影响固体超强酸的催化性能.在n(Zr)∶n(Si)=1∶3,焙烧温度550℃,焙烧时间3 h,硫酸浓度1 mol/L,陈化时间12 h的条件下,对固体超强酸的前驱体进行不同温度的预焙烧和不进行预焙烧2种工艺,其他条件不变,探究预焙烧对固体超强酸催化性能的影响.结果见表2.

表2 预焙烧对固体超强酸催化性能的影响Tab.2 Effect ofpre-calcinationon on catalytic performance ofsolid superacid
由表2可知:在200℃的预焙烧条件下固体超强酸的催化活性最高;未经预焙烧的固体超强酸催化活性稍低;随着预焙烧温度的升高固体超强酸的催化活性有降低的趋势.
2.3 H2SO4浸渍液用量对固体超强酸催化性能的影响
固体超强酸的酸性与吸附在金属离子上的SO42-的数量有关,因此硫酸溶液的用量对固体超强酸的催化性能有一定的影响.在n(Zr)∶n(Si)=1∶3,预焙烧温度200℃,焙烧温度550℃,焙烧时间3 h,硫酸浓度1 mol/L,陈化时间12 h的条件下,改变硫酸浸渍液的用量,探究其对固体超强酸催化性能的影响.结果见表3.

表3 H2SO4浸渍液用量对固体超强酸催化性能的影响Tab.3 Effect ofthe amount ofH2SO4on catalytic performance ofsolid superacid
由表3可知,m(前驱体/g)∶V(H2SO4/mL)=1∶10时,1,4-二溴丁烷收率最高;当m(前驱体/g)∶V(H2SO4/mL)=1∶14时,1,4-二溴丁烷收率没有显著的变化.考虑成本因素,选择m(前驱体/g)∶V(H2SO4/mL))=1∶10为最佳H2SO4浸渍液用量.
2.4 固体超强酸制备的正交试验设计
固体超强酸制备过程中影响因素有很多种,选取主要影响因素硅锆比例n(Si)∶n(Zr)、H2SO4浓度、焙烧温度和焙烧时间设计四因素三水平正交试验,见表4.

表4 固体超强酸制备正交试验因素水平表Tab.4 Factors and levels graph oforthogonal test for preparation ofsolid superacid
在预焙烧温度200℃,陈化时间12 h,m(前驱体/g)∶V(H2SO4/mL)=1∶10的条件下进行正交试验,结果见表5.

表5 固体超强酸制备正交试验结果Tab.5 Orthogonal test results ofsolid superacid preparation
由表5可知,R焙烧温度>Rn(Zr):n(Si)>R焙烧时间>RHSO浓度,即焙烧温度对1,4-二溴丁烷的收率的影响最大,接下来依次为硅锆比例、焙烧时间H2SO4浓度.最佳反应条件为n(Si)∶n(Zr)=1∶4,焙烧温度为550℃,焙烧时间为3 h,硫酸浓度为1.00 mol/L.
2.5 焙烧温度对固体超强催化性能的影响
由正交试验结果可知,固体超强酸催化性能的最大影响因素为焙烧温度.在预焙烧温度200℃,陈化时间12 h,m(前驱体/g)∶V(H2SO4/mL)=1∶10,n(Si)∶n(Zr)=1∶4,焙烧时间为3 h,硫酸浓度为1.00 mol/L条件下,改变焙烧温度,探究焙烧温度对固体超强催化性能的影响.结果见表6。

表6 焙烧温度对固体超强酸催化性能的影响Tab.6 Effect ofcalcination temperature on catalytic propertyofsolid superacid
由表6可知.在550℃下焙烧制备的固体超强酸催化合成的1,4-二溴丁烷收率(66.57%)最高,高于正交试验中的所有收率,即n(Si)∶n(Zr)=1∶4,焙烧温度为550℃,焙烧时间为3 h,硫酸浓度为1.00 mol/L为最佳条件,印证了正交试验的结果.
2.6 SO42-/ZrO2固体超强酸的催化效果
在预焙烧温度200℃,陈化时间12 h,m(前驱体/g)∶V(H2SO4/mL)=1∶10,焙烧温度550℃,焙烧时间为3 h,硫酸浓度为1.00 mol/L条件下,不掺杂氧化硅制备SO42-/ZrO2固体超强酸,得到1,4-二溴丁烷的收率仅为41%,可知掺杂氧化硅可以提高氧化锆的催化性能,可能是由于掺杂氧化硅增大了固体超强酸的比表面积、使载体和含S活性组分更加稳定、有效地提高固体超强酸的酸强度和总酸量[7].
2.7 固体超强酸的重复利用
对最佳工艺条件制备的固体超强酸进行反复利用,经4次催化反应后固体超强酸的催化性能明显降低.
固体超强酸的催化次数与1,4-二溴丁烷的收率关系如表7所示.

表7 固体超强酸的催化次数与1,4-二溴丁烷收率的关系Tab.7 The relationship between the catalytic times ofsolid superacid and the yield of1,4-dibromobutane
由表7可知,固体超强酸催化剂可重复利用,但催化剂有一定的使用寿命,重复使用3次之后催化剂基本上失活.试验证明将失活后的固体超强酸用硫酸浸渍之后经过焙烧再重复利用其催化1,4-二溴丁烷,收率可达64.73%.
2.8 固体超强酸及1,4-二溴丁烷的结构表征
2.8.1 固体超强酸的红外分析对3种不同焙烧温度下制备的固体超强酸进行红外光谱测试.将干燥后的溴化钾与制备的固体超强酸混合研磨,在压片机上压制成透明薄片,在红外光谱仪上进行测定,测定波长范围为4000~4 00 cm-1,得到的红外光谱如图1所示.

图1 不同焙烧温度制备的固体超强酸的红外光谱Fig.1 Infrared spectra ofsolid superacid prepared at different calcination temperatures
由图1可以看出,3 400 cm-1吸收峰为吸附在固体超强酸酸性中心上水的—OH伸缩振动吸收峰,相比较而言在550℃下焙烧的固体超强酸吸水性更强一些;在760 cm-1处出现吸收峰,说明焙烧后的固体超强酸晶型为单斜晶[8];3种样品都在1 200 cm-1有强吸收,说明3种条件下焙烧的固体超强酸中的SO42-与金属氧化物之间是以双配位键相作用的,但是吸收峰集中在1 200 cm-1以下,则说明本方法制备的固体超强酸中SO42-与金属氧化物之间主要是以桥式双配位键相结合的.
2.8.2 1,4-二溴丁烷的红外分析对合成的1,4-二溴丁烷采用溴化钾压片法进行红外光谱测试.测定波长范围为400~4 000 cm-1,得到的红外光谱图如图2所示.
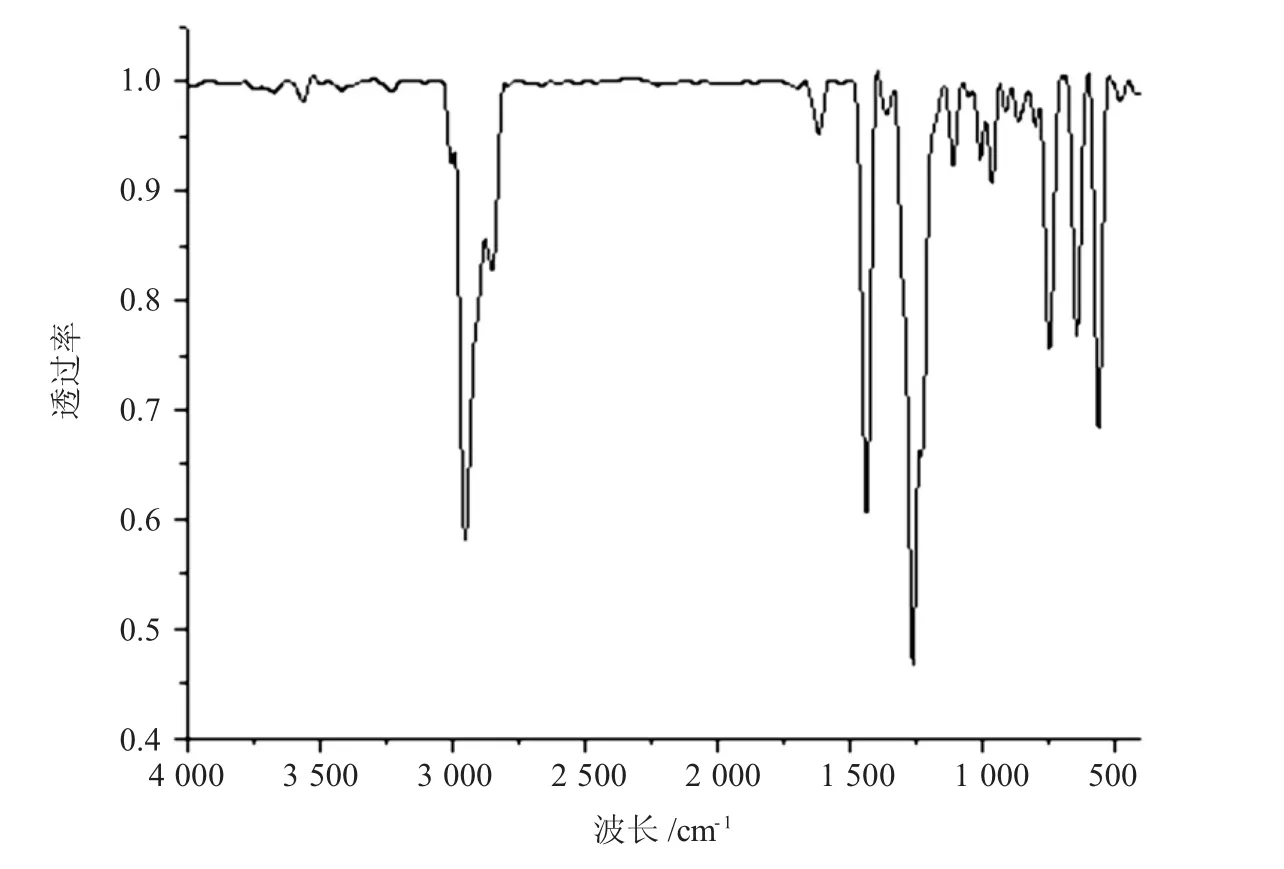
图2 合成的1,4-二溴丁烷红外光谱Fig.2 Infrared spectra of1,4-dibromobutane synthesized
由图2可知,在2 956.62 cm-1处出现较强的吸收峰带,该吸收峰为亚甲基(-CH2-)的强伸缩振动带;在1 440.66 cm-1处出现中等强度伸缩振动吸收带,为亚甲基(-CH2-)的吸收带;在1 265.28 cm-1附近为-CH2-面内弯曲振动产生的红外吸收,为-CH2-Br;而在749~563 cm-1出现的峰则为C-Br伸缩振动吸收带.这与标准红外谱图中,在2961cm-1处出现较强的吸收峰带,在1446cm-1处出现吸收带,在1 288 cm-1与656 cm-1出现吸收峰相符.从而可证明所合成的产品为1,4-二溴丁烷.
2.8.3 固体超强酸的比表面积分析催化剂的比表面积对催化剂的活性大小具有非常重要的影响,利用全自动BET氮吸附法比表面积测定仪分析测量不同焙烧温度下固体超强酸的比表面积,结果见表8.

表8 不同焙烧条件下固体超强酸的比表面积Tab.8 The specific area ofsolid superacid under different calcination temperature
由表8可知,焙烧温度与固体超强酸的比表面并非线性关系,其催化活性也并非随固体超强酸比表面积的降低而降低.
在焙烧过程固体超强酸表面的H2SO4随着温度的升高逐渐被除去,金属氧化物由无定型转化为单斜晶,离子型的S=O键转变为共价键形式的S=O键,但是随着焙烧温度的进一步升高,催化剂表面的SO42-被还原为SO2从金属氧化物表面脱离,造成固体超强酸酸性中心减少,酸强度降低,催化活性降低[9].这也是焙烧温度是固体超强酸催化活性的最大影响因素的原因之一.
综上所述,焙烧温度为550℃时固体超强酸的比表面积最大,尽管其金属氧化物表面吸附的SO42-数量并不是最多的,但是最大的比表面积与相对较多的酸性中心,使得其催化活性最大.
2.8.4 1,4-二溴丁烷的气相色谱分析气相色谱测试条件:SE-30毛细管,FID检测器,程序升温,初始温度50℃,初温保持时间1 min,升温速率10℃/min,终止温度200℃,终温保持时间5 min,积分方法为面积归一法,分析结果见表9.
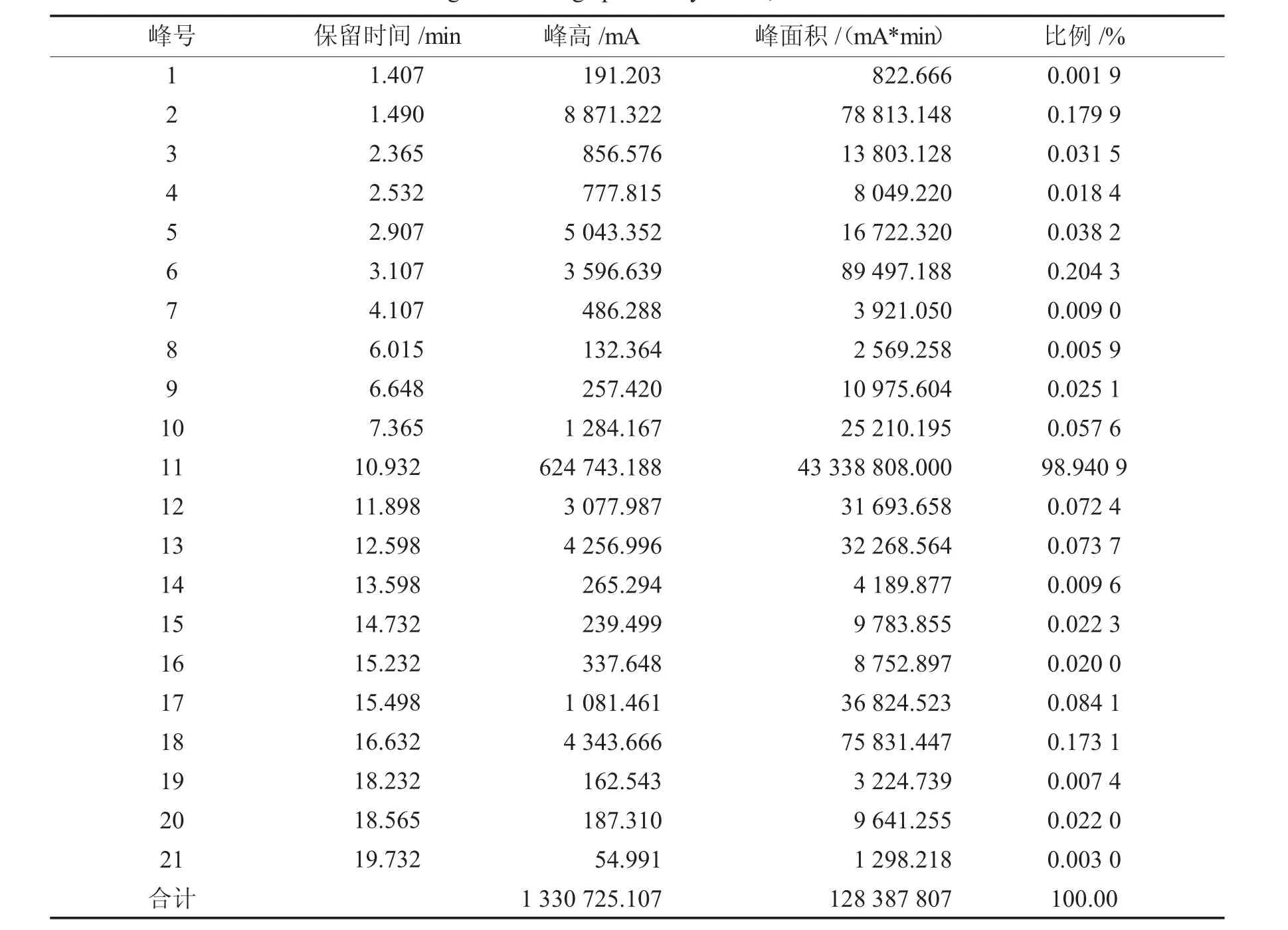
表9 1,4-二溴丁烷的气相色谱分析Tab.9 The gas chromatographic analysis of1,4-dibromobutane
由气相色谱图可得,在设定的测试条件下1,4-二溴丁烷保留时间为10.932 min,纯度在98.9409%. 2.8.5 1,4-二溴丁烷的折光率测试经测试样品折光率为1.516 9,此数值与文献值(1.519 0)稍有误差.由于1,4-二溴丁烷在光照条件下容易分解,产品里面还残留有未反应的1,4-丁二醇(折光率1.445~1.446)使得产物的纯度降低(纯度:98.940 9%),因此折光率略小于文献值.
3 结论
本论文制备了SO42-/ZrO2-SiO2固体超强酸,并用制备的固体超强酸作为催化剂合成1,4-二溴丁烷,用1,4-二溴丁烷得收率为指标,确定了制备固体超强酸得最佳工艺:n(Zr)∶n(Si)=1∶4,陈化时间12 h,预焙烧温度200℃,焙烧温度550℃,焙烧时间3 h,H2SO4浓度1.00 mol/L,m(前驱体/g)∶V(H2SO4/mL)=1∶10,最佳条件下制备的固体超强酸催化合成1,4-二溴丁烷的和收率可达到66.57%,所制备的固体超强酸虽然具有一定的催化作用,但是催化能力却小于浓硫酸.但是固体超强酸催化剂具有对设备无腐蚀性、对环境污染小的优势,并且可重复利用,固体超强酸催化剂的催化活性随着重复使用次数的增加而降低,重复使用3次之后催化剂基本上失活,将失活的固体超强酸用硫酸促进剂浸渍、焙烧后将重新恢复活性.
[1]周国斌,陈慧宗,徐景士,等.固体超强酸及其催化的有机化学反应[J].江西化工,2001(1):3-8.
[2]朱正峰.固体超强酸的制备、表征及其在酯化反应中的应用[D].呼和浩特:内蒙古工业大学,2007.
[3]汪洋,朱光宇,于贺,等.SO42-/MxOy型固体超强酸的制备及其改性研究[J].化学与生物工程,2013,30(6):22-71.
[4]韩周冰,张刚,陈先明,等.SO42-/MxOy型固体超强酸及其在酯化中的应用进展[C]//第四届全国工业催化技术及应用年会论文集,2007:4.
[5]唐新硕,王新平,金松寿.SO42-/ZrO2型超强酸酸中心形成机理研究[J].中国科学,1994,24(6):584-595.
[6]高占先.有机化学[M].北京:高等教育出版社,2008:300-301.
[7]夏勇德,华伟明,高滋.Al促进SO24-/MxOy(M=Zr,Ti,Fe)固体超强酸的研究[J].化学学报,2000,58(1):86-91.
[8]袁荞龙,周风华,吴树森.ZrO2-SiO2纳米复合微粒的制备与性能[J].华东理工大学学报(自然科学版),2007,33(4):489-495.
[9]YANGH,LUR,ZHAOJ,et al.Sulfated binaryoxide solid superacids[J].Materials Chemistryand Physics,2003,80(1):68-72.
(责任编辑:卢奇)
Research on the synthesis of SO42-/ZrO2-SiO2solid superacid
XU Fenghua,ZHAI Zhuhe,TIAN Mengchao,LI Aimei,ZHAO Ya’nan
(Zhongyuan UniversityofTechnology,Zhengzhou 450007,China)
TQ426.6
A
1008-7516(2017)04-0047-07
10.3969/j.issn.1008-7516.2017.04.009
2017-05-10
徐凤华(1991—),女,河南鹿邑人,硕士.主要从事有机合成与绿色染整技术研究.
田孟超(1962—),男,河南伊川人博士,教授.主要从事有机合成及精细化工研究.
猜你喜欢
Acta Mathematica Scientia(English Series)(2021年1期)2021-04-08 12:52:22
中国茶叶加工(2020年3期)2020-10-21 08:06:48
石油石化绿色低碳(2019年6期)2019-01-14 01:16:20
材料科学与工程学报(2016年1期)2017-01-15 13:33:52
当代化工研究(2016年7期)2016-03-20 16:21:54
合成化学(2015年10期)2016-01-17 08:56:29
电源技术(2015年9期)2015-06-05 09:36:06
化学反应工程与工艺(2015年3期)2015-04-16 03:06:19
应用化工(2014年11期)2014-08-16 15:59:13
应用化工(2014年1期)2014-08-16 13:34:08
