
超高效液相色谱-串联质谱法同时检测鸡肉中氯霉素、四环素、金霉素和土霉素
2017-09-22 05:43梅英杰史新宇董瑾朱西雷张贞理李德刚
食品与发酵工业 2017年8期
梅英杰,史新宇,董瑾,朱西雷,张贞理,李德刚
1(山东理工大学 化学工程学院,山东 淄博,255000) 2(淄博市食品药品检验研究院,山东 淄博,255000) 3 (淄博市产品质量监督检验所,山东 淄博,255000)
超高效液相色谱-串联质谱法同时检测鸡肉中氯霉素、四环素、金霉素和土霉素
梅英杰1,2,史新宇3,董瑾2,朱西雷2,张贞理2,李德刚1*
1(山东理工大学 化学工程学院,山东 淄博,255000) 2(淄博市食品药品检验研究院,山东 淄博,255000) 3 (淄博市产品质量监督检验所,山东 淄博,255000)
采用固相萃取法对鸡肉中氯霉素、四环素、金霉素和土霉素残留同时提取、净化、富集,并建立了用超高效液相色谱-串联质谱法对4种抗生素定性定量检测的分析方法。前处理部分采用含0.1%三氯乙酸的乙腈为提取溶剂,饱和了乙腈的正己烷溶液去除脂肪,超声波萃取2次;采用HLB固相萃取柱净化;色谱部分采用BEH C18色谱柱(2.1 mm×50 mm,1.7μm),乙腈-0.2%甲酸水(含5 mmol/L乙酸铵)为流动相进行梯度洗脱。在最优条件下氯霉素、四环素、金霉素和土霉素在相应浓度范围内,线性良好,相关系数R2均大于0.999,其检出限分别为0.1、3.2、16.0、9.0μg/kg,日内、日间相对标准偏差(n=5)分别小于5.0%和10.0%,平均回收率为80.1%~92.5%。
氯霉素;四环素;固相萃取;超高效液相色谱-串联质谱
抗生素添加到饲料中,能有效提高饲养物的存活率和生长速度[1]。随着肉制品的需求不断加大,以及养殖过程中畜禽疾病频发,诊断难度增大,导致养殖业抗生素药物滥用、过量使用现象日趋严重[2]。人们食用含抗生素残留的肉制品后,抗生素残留物会在人体蓄积,会对人体健康造成有害影响[3]。另一方面,这些抗生素残留会伴随动物尿液、粪便等转移到外部环境中,而这些排泄物中有些抗生素成分还未失活,在环境中积累、富集,进而对土壤的生态环境和动植物体造成影响。
目前,动物性食品中氯霉素与四环素类的检测方法主要有酶联免疫法[3]、微生物学法[4]、气相色谱-质谱联用法[5]、液相色谱法[6]与液相色谱-质谱联用法[7-9]等。酶联免疫法操作简便,特异性强,常用于批量样品的快速筛选,但定量准确度较差;微生物学法易于操作,成本低,但检测周期长,特异性差,干扰多;气相色谱-质谱联用法检测时需进行衍生化,操作繁琐费时;液相色谱法采用光谱检测时定性手段较单一,易出现假阳性结果;液相色谱-质谱联用法(LC-MS)通过液相色谱将目标物分离,以质谱仪为定性定量手段,可有效避免假阳性结果。超高效液相色谱-串联质谱技术(UPLC-MS/MS)相比较传统的LC-MS,显著提高了检测的灵敏度、准确度、重复性,能更好地适应现代检测技术对高通量、高精度的需要,在抗生素残留分析应用中发展迅速[10-13]。本文以固相萃取为前处理手段,通过UPLC-MS/MS分析,建立一套完整的分析方法,实现对鸡肉中氯霉素、四环素、金霉素、土霉素同时提取、同时检测。
1 材料与方法
1.1实验仪器、试剂
1.1.1 实验仪器
电子天平(MS205DU),METTLER TOLEDO公司;电子天平(ML204/02),梅特勒-托利多仪器(上海)有限公司;氮吹仪(HN200),海能仪器有限公司;固相萃取仪(JTCQ-24B),杭州聚同电子有限公司;高速冷冻离心机(TGL-20M),长沙湘仪离心机仪器有限公司;超声波清洗仪(SK2200H),上海科导超声仪器有限公司;超纯水仪(Integral 5),MILLIPORE公司;超高效液相色谱-串联质谱仪(ACQUITY Quattro Premier XE),美国沃特斯公司。
1.1.2 实验试剂
氯霉素标准物质(≥99.8%),中国计量科学研究院;四环素、金霉素、土霉素单标溶液(100 μg/mL),农业部环境保护科研监测所;乙酸铵(分析纯)、三氯乙酸(分析纯),天津市福晨化学试剂厂;无水Na2SO4(分析纯),莱阳市经济技术开发区精细化工厂;正己烷(HPLC级)、甲酸(HPLC级),天津市科密欧化学试剂有限公司;甲醇(HPLC级)、乙腈(HPLC级),Fisher公司。
1.2标准溶液的配制
1.2.1 标准储备液的配制
准确称取氯霉素10 mg于100 mL烧杯中,加入约30 mL甲醇溶解,溶解较慢时可超声5min加速溶解,待标准物质完全溶解后,转入100 mL棕色容量瓶中,并用甲醇定容至100 mL,该标准储备液浓度为100 mg/L,配制好后,放入4 ℃冰箱中保存。
1.2.2 混合标准使用液的配制
分别准确移取浓度均为100 mg/L的氯霉素、四环素、金霉素、土霉素各1.0 mL于10 mL棕色容量瓶中,加甲醇至刻度,配制成单标中间液浓度为10 mg/L,配制好后放入4℃冰箱中保存。
用甲醇稀释分别配制不同浓度系列的混合标准使用液,经0.2μm微孔滤膜过滤后待测。
1.3流动相的配制
0.2%甲酸水溶液(含5 mmol/L的乙酸铵):称取0.38 g乙酸铵于150 mL烧杯中,加50 mL超纯水溶解,加入2 mL甲酸,转移至1 000 mL容量瓶,加水定容至刻度。
乙腈:色谱纯。
1.4样品前处理
1.4.1 样品的制备
购买市售白条鸡,取肌肉部分。用高速组织捣碎机将样品打碎后,将样品混匀,放入干净的样品盘中,保存备用。
1.4.2 提取液的配制
0.1%的三氯乙酸溶液:称取三氯乙酸1.0g加水定容至1 L。乙腈:色谱纯。乙腈饱和过的正己烷:取100 mL正己烷于分液漏斗中,加入100 mL乙腈混合至分层,取上层溶液备用。
1.4.3 提取过程
称取5.00 g样品于50 mL聚四氟乙烯离心管中,加入5 mL的0.1%三氯乙酸溶液,10 mL乙腈,振荡混匀1 min后,超声提取10 min,10 000 r/min离心5 min,上清液转移至另一洁净离心管中,向残留物中再加入10 mL乙腈,超声提取10 min,10 000 r/min离心5 min,合并上层清液,加入5.0 g无水Na2SO4,加入5 mL乙腈饱和的正己烷溶液,涡旋混合1 min后,静置5 min,取乙腈溶液于室温下氮气吹干,加入0.1%三氯乙酸溶液2 mL复溶,备用。
1.4.4 样品净化
将Oasis HLB固相萃取柱以3 mL甲醇和3 mL超纯水分别纯化,将提取液注入固相萃取柱(通过固相萃取柱的流速不宜过快,基本控制在1 min/mL),以6 mL 10%甲醇水溶液淋洗小柱,抽干,加入6 mL V(甲醇)∶V(乙酸乙酯)=15∶85洗脱,抽干5 min,收集洗脱液于室温下氮气吹干,用1 mL流动相溶解,过滤备用。
1.5测定条件
1.5.1 液相部分
色谱柱:ACQUITY UPLC BEH C18色谱柱 1.7 μm、2.1mm×50 mm ;进样量:10μL;柱温: 40℃;
流动相: A为0.2%甲酸水溶液(含5 mmol/L乙酸铵),B为乙腈;流速0.20 mL/min;梯度条件:0~5 min:A 90%~60%;5~6 min:A 60%~5%,A 5%保持2 min;8.1 min:A 90%。
1.5.2 质谱条件
调整质谱参数,得到如表1所示最优质谱条件。
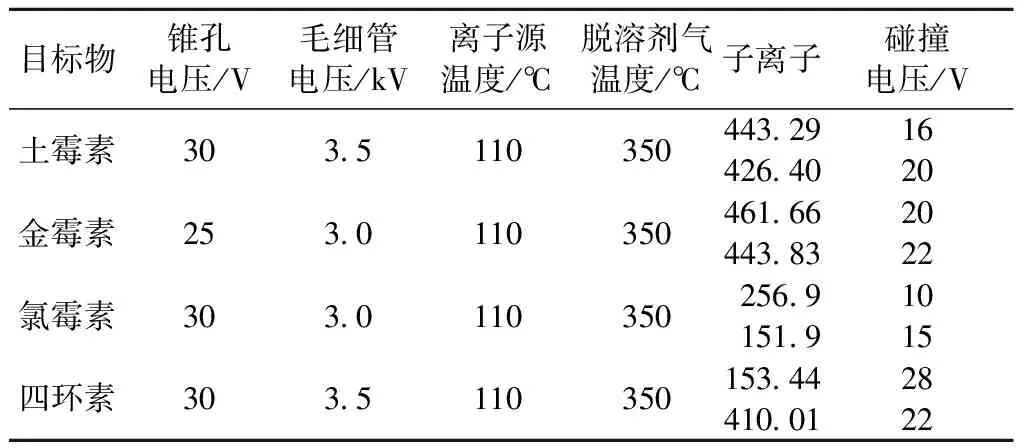
表1 质谱条件
2 结果与讨论
2.1前处理条件优化
2.1.1 提取溶剂的选择
从生物样品中提取氯霉素和四环素类比较复杂,生物基质中存在的蛋白质会影响目标物的提取效率,因此在提取时考虑用强酸溶液或酸性溶液对蛋白质进行分离。但是,在较强酸性环境中(pH<2.0),四环素类药物会降解为脱水物,发生分解。因此,提取时宜选择弱酸性溶剂。常用的弱酸性溶剂有EDTA-Mcilaine缓冲液(pH 4.0)、酸化乙腈、酸化乙腈-甲醇等。另外,三氯乙酸溶液、柠檬酸盐缓冲液等也可用于样品的提取和沉淀蛋白质。张鑫等采用EDTA-Mcilaine缓冲液对鸡肉中四环素类抗生素进行了提取,经固相萃取净化后,用UPLC-MS/MS定性定量检测[14];洪波等采用酸化乙腈对水产品中四环素进行了提取,用液相色谱进行了定量检测[15];谢寒冰等采用酸化乙腈-甲醇对猪肉中四环素类等多种抗生素进行提取,并用液相色谱-四极杆飞行时间质谱进行了定性定量检测[16]。氯霉素性质稳定,较易提取。提取动物组织中氯霉素时,常用到乙酸乙酯和乙腈溶液。冯雷等以乙酸乙酯为提取剂,对禽肉中的氯霉素进行了提取,用液相色谱-质谱联用法进行了定性定量检测[17];陈剑刚等以乙腈为提取液对虾肉中氯霉素提取后,用液相色谱-串联质谱法进行了定性定量检测[18]。本文选用0.1%三氯乙酸-乙腈提取体系,在弱酸性条件下有效提取氯霉素、四环素类抗生素残留,并同时沉淀样品中的蛋白质,效果较好。由于动物组织中存在较多的游离脂肪,会进入到萃取液中,影响萃取效果,因此需对提取溶液进行脱脂处理,选用乙腈饱和的正己烷进行脱脂处理。
2.1.2 提取方式的选择
在提取过程中,提取方式对目标物的提取速度和完全程度存在一定影响。提取时间在一定程度上决定了提取的完全程度,提取时间长,提取的完全程度高,但提取效率低;提取时间缩短,虽然有助于提取效率提高,但可能会造成提取不完全。实验室常用的提取方式有:静置提取、振荡提取和超声波提取等。实验中,对静置提取,振荡提取和超声波提取分别作了比较。发现3种提取方式分别提取时,短时间内,超声波提取的回收率最高,振荡提取次之,静置提取最低,这是由于在短时间内静置提取和振荡提取对样品的浸润和渗透能力不足,造成提取效果的降低。而利用超声波提取时,由于超声波的“空化”作用,加速了目标物的溶出,提取效率提高。因此,在实验中选取超声波提取作为目标物提取方式。
2.1.3 固相萃取条件的选择与优化
固相萃取柱选用Oasis HLB柱,其键合相为N-乙烯吡咯烷酮和亲脂性的二乙烯基苯聚合物,其回收率好于普通的C18小柱。固相萃取过程中,洗脱液的选择对目标物回收率有重要影响。由于测试样品为动物组织,基质复杂,为提高目标物的洗脱完全率,本文选用了甲醇/乙酸乙酯混合液作为洗脱剂,分别采用V(甲醇)∶V(乙酸乙酯)=5∶95、10∶90、15∶85、20∶80洗脱液对提取后的基质加标样品进行洗脱并测定,通过比较如图1所示不同洗脱液对应的目标物回收率,最终确定V(甲醇)∶V(乙酸乙酯)=15∶85 作为洗脱液。
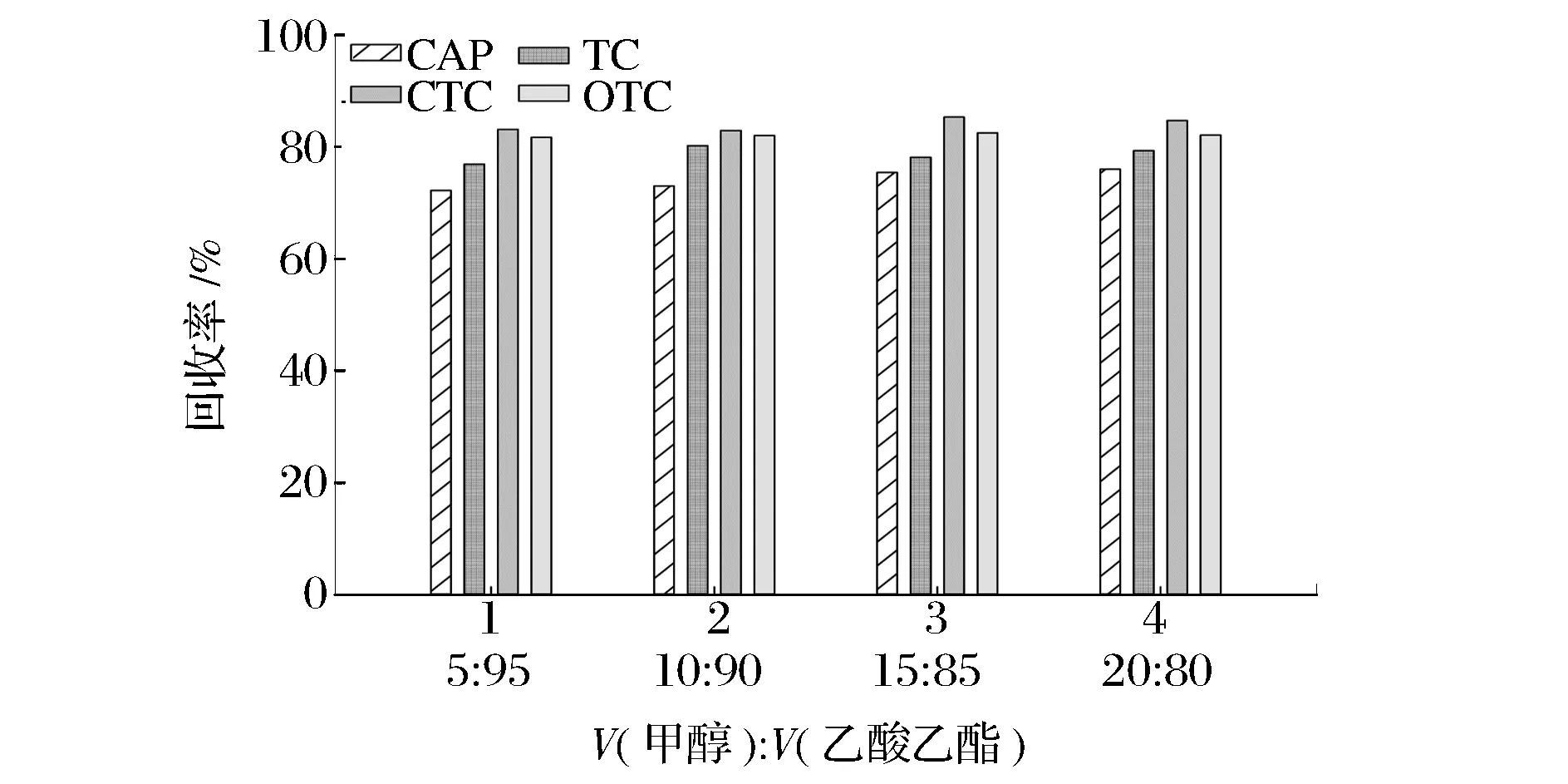
图1 不同洗脱液对4种目标物回收率的影响Fig.1 Effect of differenteluent on recovery
2.2分析条件优化
2.2.1 流动相的确定
液相色谱-质谱联用分析中,流动相一般选择黏度低,通用性好的溶液。实验中分别选用乙腈-水、乙腈-0.2%甲酸水、乙腈-0.2%甲酸水溶液(含5 mmol/L乙酸铵)做流动相进行试验。试验表明,3种流动相体系对氯霉素的保留与响应影响不大,但四环素、金霉素、土霉素灵敏度很差,无法满足要求。采用乙腈-0.2%甲酸水体系时,四环素、金霉素灵敏度满足要求,但土霉素灵敏度较低。采用乙腈-0.2%甲酸水溶液(含5 mmol/L乙酸铵)体系时,4种抗生素响应均能满足要求,故本次研究采用乙腈-0.2%甲酸水溶液(含5 mmol/L乙酸铵)作为流动相。
2.2.2 电离模式的选择
分别配制浓度为1μg/mL的氯霉素、四环素、金霉素、土霉素的标准溶液,以流动注射方式进样,流速为20 μL/min,分别在正离子模式(ESI+)和负离子模式(ESI-)下进行全扫描,扫描完成后,分别比较正、负离子模式下4种抗生素的响应值,结果表明,氯霉素在负离子模式下响应正常,正离子模式下基本无响应;四环素、金霉素、土霉素在正离子模式下响应正常,在负离子模式下基本无响应,故采集氯离子宜选用负离子模式,四环素等采用正离子模式采集。本文采用正负离子双通道同时采集模式实现对这4种抗生素同时检测。
2.2.3 母离子与子离子的确定
本文采用三重四级杆串联质谱仪,通过调整质谱参数,得到化合物的一级质谱图,得到目标物的分子量信息,确定目标物的母离子。氯霉素、四环素、金霉素、土霉素母离子分别为m/z 320.9、444.97、479.17、461.00,与相关标准记载及文献报道一致[7-8,19-20]。
配制氯霉素、四环素、金霉素、土霉素的标准液,根据前述确定的色谱、质谱条件进行测定,得到如图2所示4种抗生素的多反应监测色谱图。
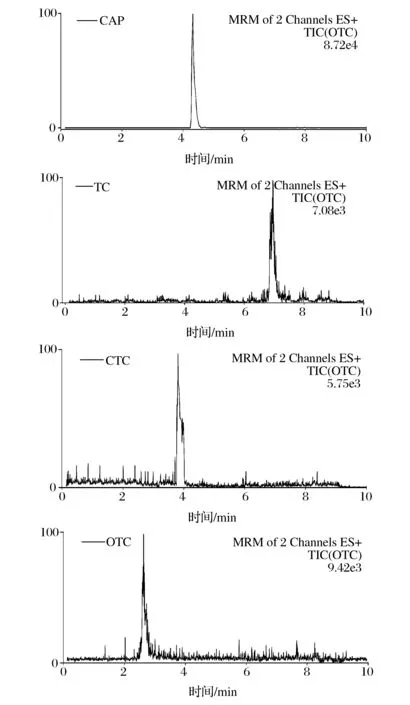
图2 氯霉素、四环素、金霉素、土霉素多反应监测色谱图Fig.2 Chromatograms ofChloramphenicol,tetracycline,chlortetracycline,oxytetracycline in multiple reaction monitoring (MRM) mode
从图2可以看出,在最优条件下,4种抗生素分离度较好,能达到基线分离,且峰形能够满足实验要求,四种目标物能在10 min内完成分离。在正负离子双通道同时采集条件下,可实现四种目标物的同时检测。4种目标物的保留时间、母离子、子离子信息如表2所示。
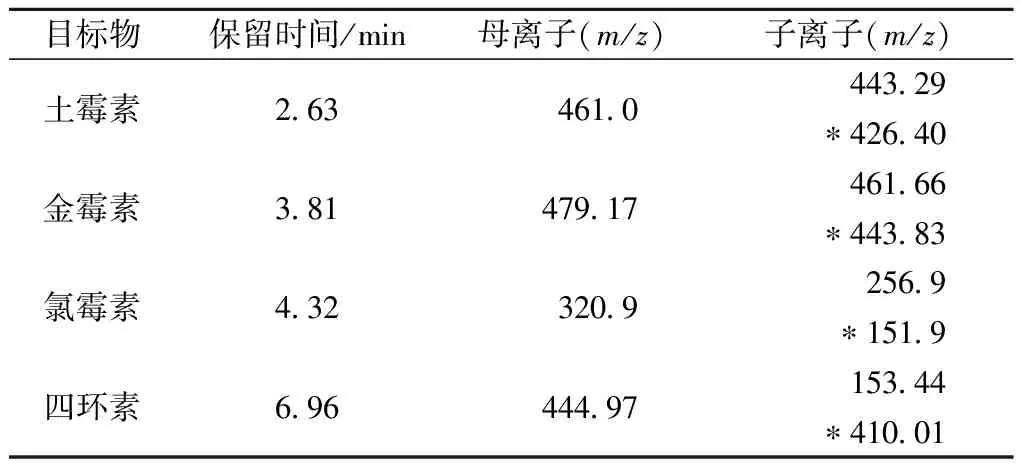
表2 四种目标物的质谱信息
注:*定量离子。
本文还研究了基质效应对测定结果的影响,研究发现,标准溶液和空白基质加标溶液的色谱图在出峰时间、基线噪声、响应值等方面无明显不同,这说明样品经前期提取净化后,基质效应对检测结果基本无影响。
2.3线性范围
在0.2~500 μg/L内配制氯霉素、四环素、金霉素、土霉素的混合标准使用液,按照本研究确定的仪器条件进行测定,以标准溶液浓度为横坐标(Xμg/L),峰面积为纵坐标(Y),拟合一次线性方程,分别得到四种目标物的一次线性回归方程,见表3。

表3 目标物的线性回归方程
2.4方法的检出限和定量限
称取5.0 g空白基质样品,采用基质加标的方法添加4种抗生素,按本文确定的前处理方法和仪器条件进行检测,按照3倍基线噪声(S/N=3)计算方法检出限,按照10倍基线噪声(S/N=10)计算方法定量限,配制检出限、定量限等同浓度标准溶液,并经仪器检测验证。方法检出限及定量限结果如表4所示。

表4 目标物的检出限及定量限
2.5方法回收率与精密度
采用基质加标方式,配制混合标准溶液,按照本文确定的仪器条件,考察方法日内精密度(同一天内连续进样5次,n=5)和日间精密度(每天进样1次,连续5天,n=5),得到3种浓度下4种抗生素的日内和日间相对标准偏差(RSD),结果见表5。结果表明4种抗生素的日内、日间相对标准偏差均小于5.0%和10.0%。3个浓度加标回收率在80.1%~92.5%之间。
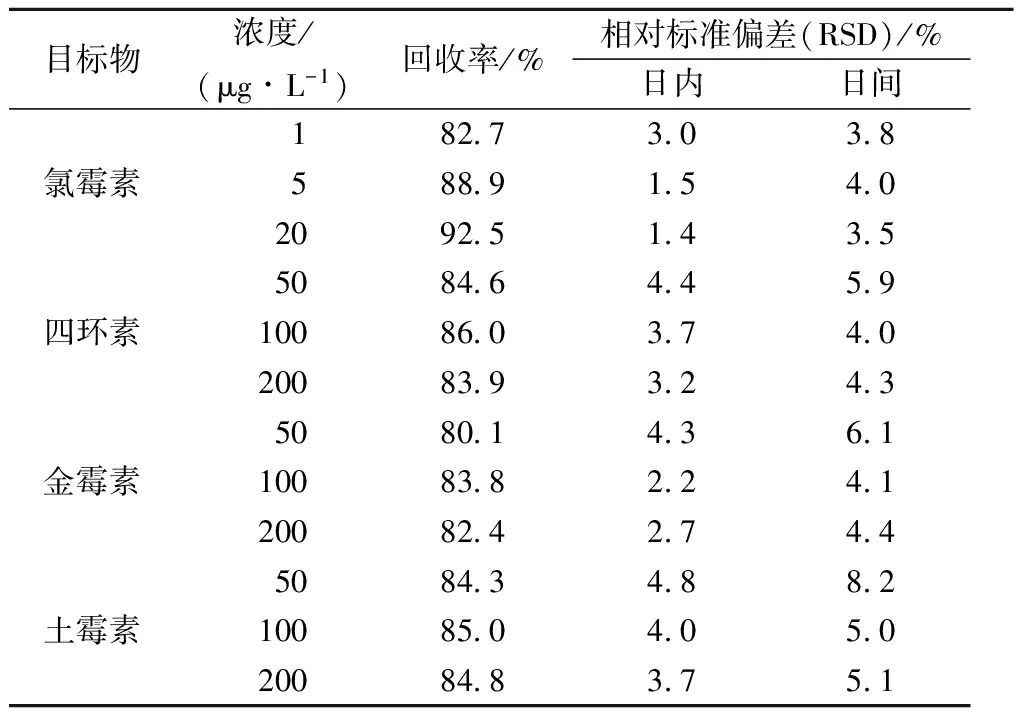
表5 回收率与日内和日间精密度(n=5)
2.6实际样品的测定
按照本文上述建立的样品前处理方法对10份鸡肉样品进行处理,在最优仪器条件下,对鸡肉样品进行定性定量分析。其中,1份鸡肉样品中只检出土霉素,定量结果为31.2 μg/kg,其多反应监测色谱图如图3所示;1份鸡肉样品中检出土霉素和氯霉素,定量结果分别为67.3 μg/kg和6.8 μg/kg,其多反应监测色谱图如图4所示。
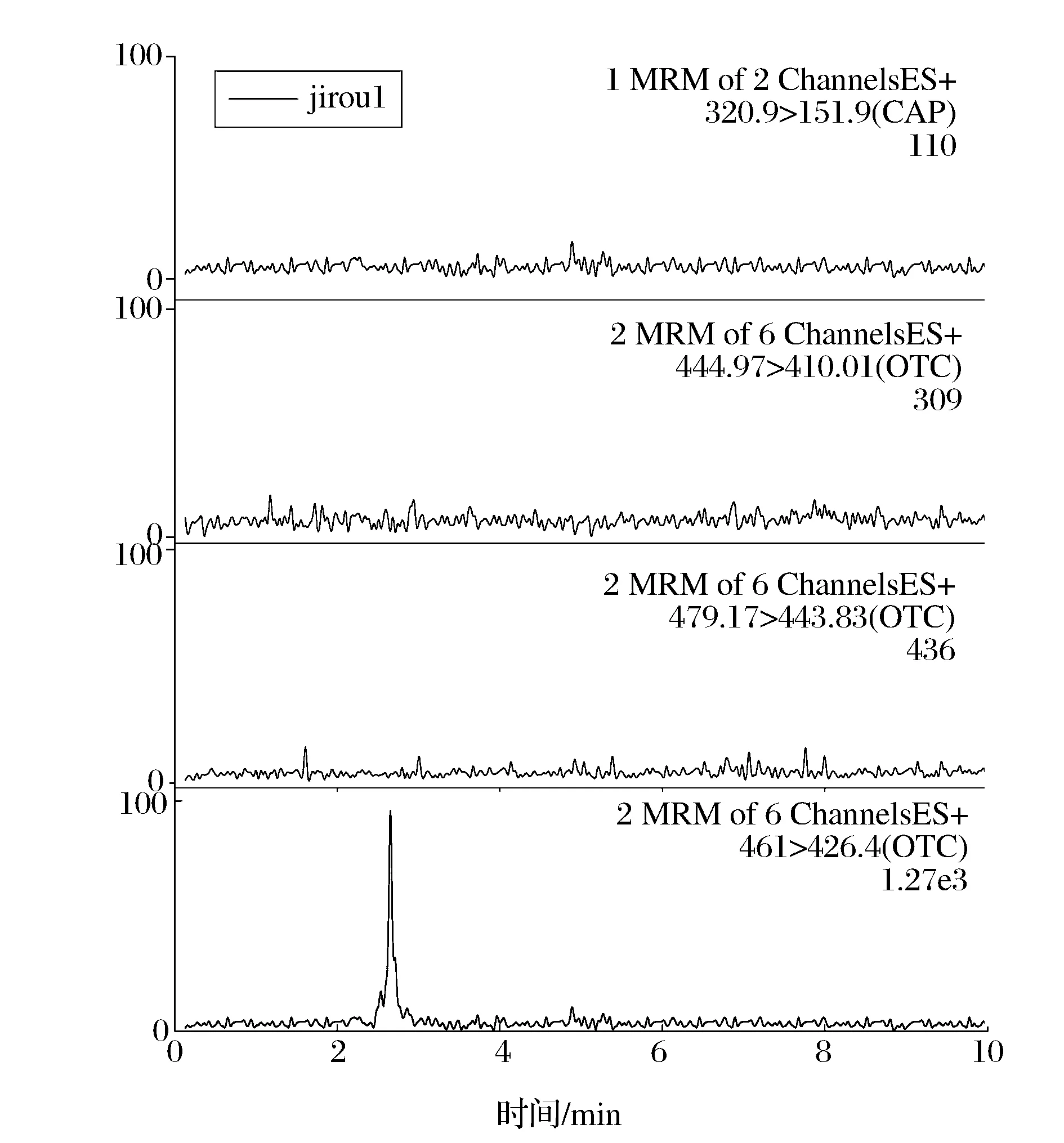
图3 阳性样品1多反应监测色谱图Fig.3 MRM chromatograms of positive sample 1
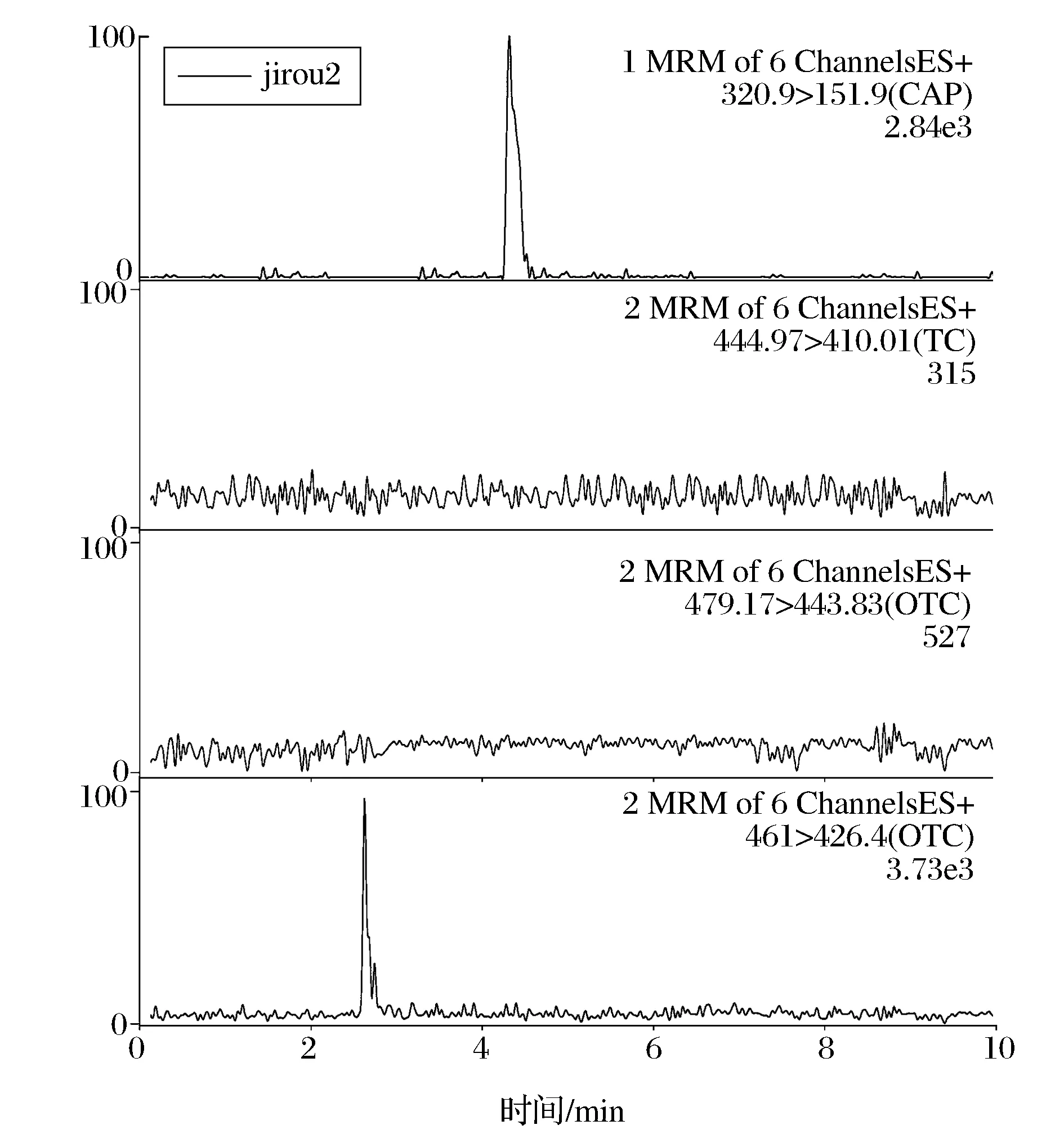
图4 阳性样品2多反应监测色谱图Fig.4 MRM chromatograms of positive sample 2
3 结论
通过优化样品前处理过程、色谱条件中的色谱柱、流动相组成及配比、柱温以及质谱条件中的锥孔电压、毛细管电压、离子源温度、脱溶剂气温度等因素,确立了鸡肉中氯霉素、四环素、金霉素、土霉素抗生素残留的UPLC-MS/MS分析方法。4种目标物在相应浓度范围内线性良好,相关系数(R2)为0.999 2~0.999 6,检出限分别为0.1、3.2、16.0、9.0 μg/kg。
[1] 陈燕凌,王志强.畜禽专用性抗生素饲料添加剂[J].兽药与饲料添加剂,2001,6(3):22-24.
[2] 李书云,蒙超林,全宏怀.柳州市场猪肉、鸡肉和牛奶的抗生素残留的调查报告[J].广西畜牧兽医,2001,17(6):12-14.
[3] CHENY N,KONG D Z,LIU L Q,et al.Development of an ELISA and immunochromatographic assay for tetracycline,oxytetracycline,and chlortetracycline residues in milk and honey based on the class-specific monoclonal antibody[J].Food Anal Methods,2016,9:905-914.
[4] 林修光,寇运同,贾臻,等.用标准纸片法快速测定鸡肉中四环素族残留量[J].口岸卫生控制,2001,6(3):19-20.
[5] 李鹏,邱月明,蔡慧霞,等.气相色谱-质谱联用法测定动物组织中氯霉素、氟甲砜霉素和甲砜霉素的残留量[J].色譜,2006,24(1):14-18.
[6] 时小艳.高效液相色谱测定加工肉制品中四环素族抗生素的含量[J].食品工业,2015(9):282-284.
[7] 周鑫,李明,张鑫,等.高效液相色谱-串联质谱法测定鸡肉中的喹诺酮类和四环素类药物残留[J].畜牧与兽医,2015,47(11):19-22.
[8] 田艳玲,王浩.液相色谱-串联三重四极杆质谱测定牛奶中的氯霉素残留量[J].食品研究与开发,2011,32(3):125-127.
[9] BOISON J O,LEE S,MATUS J.A multi-residue method for the determination of seven polypeptide drug residues in chicken muscle tissues by LC-MS/MS.[J].Analytical & Bioanalytical Chemistry,2015,407(14):1-14.
[10] 张鑫,吴剑平,李丹妮,等.UPLC-MS/MS检测七种动物源食品中四环素类药物残留量的研究[J].中国兽药杂志,2015(12):36-42.
[11] 张伟,彭麟,江善祥.超高效液相色谱-串联质谱法测定畜禽饲料中氯霉素类药物含量[J].南京农业大学学报,2015,38(3):453-458.
[12] 刘兆峰,钟启升.超高效液相色谱三重四极杆质谱联用法测定蜂蜜中5种四环素类抗生素的探讨[J].分子诊断与治疗杂志,2015,7(6):416-421.
[13] LI Y H,LIU Z Q,CHEN Y M,et al.Determination ofoxytetracycline,tetracycline and chlortetracycline residues in meat by UPLC-ESI-MS/MS[J].Chinese Journal of Health Laboratory Technology,2008,18(1):96-98.
[14] 张鑫,吴剑平,李丹妮,等.UPLC-MS/MS检测七种动物源食品中四环素类药物残留量的研究[J].中国兽药杂志,2015,49(12):36-42.
[15] 洪波,曾春芳,高峰,等.高效液相色谱—紫外法测定水产品中四环素类、喹诺酮类抗生素残留[J].湖南农业科学,2013(21):81-84.
[16] 谢寒冰,王毅刚,周明莹,等.液相色谱-四极杆飞行时间质谱法筛查猪肉中5类26种药物残留[J].分析测试学报,2014,33(4):373-379.
[17] 冯雷,尹丽珠,孙文通,等.禽畜肉中氯霉素残留量的液质联用分析方法[J].食品科学,2010,31(4):243-245.
[18] 陈剑刚,张艳,白艳玲,等.同位素内标稀释液相色谱-串联质谱法测定虾中氯霉素、甲砜霉素和氟甲砜霉素残留量[J].中国卫生检验杂志,2013(7):1670-1673.
[19] 中华人民共和国国家质量监督检验检疫总局.GB/T 21317—2007动物源性食品中四环素类兽药残留量检测方法液相色谱-质谱/质谱法与高效液相色谱法[S].北京: 中国标准出版社,2007.
[20] 中华人民共和国农业部.动物源食品中氯霉素残留量的测定高效液相色谱-串联质谱法[S].北京:中国农业出版社,2006.
Determinationofchloramphenicol,tetracycline,chlortetracyclineandoxytetracyclineinchickenbyultraperformanceliquidchromatography-tandemmassspectrometry
MEI Ying-jie1,2,SHI Xin-yu3,DONG Jin2,ZHU Xi-lei2,ZHANG Zhen-li2,LI De-gang1*
1 (College of Chemical Engineering,Shandong University of Technology,Zibo 255000,China) 2 (Zibo Institute for Food and Drugcontrol,Zibo 255000,China) 3 (Zibo Institute of Product Quality Supervision & Inspection,Zibo 255000,China)
A solid phase extraction column clean-up and ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method was developed for simultaneous determination of chloramphenicol,tetracycline,chlortetracycline and oxytetracycline in chicken products.In the pre-treatment process,the sample was extracted with 0.1% trichloroacetic acid-acetonitrile and ultrasonic treatment twice,and fat was removed by acetonitrile saturated n-hexane.HLB solid phase extraction column was used to purify.BEH C18(2.1 mm×50 mm,1.7 μm) column was used and acetonitrile-0.2% formic acid (containing 5mmol/L ammonium acetate) was the gradient eluent.Under the above optimized conditions,chloramphenicol,tetracycline,chlortetracycline and oxytetracycline had a good linear correlation in the corresponding concentration ranges (R2> 0.999).The detection limits of four substances were 0.1,3.2,16.0,9.0 μg/kg,respectively.Intra-day and inter-day relative standard deviations (n= 5) were less than 5.0% and 10.0% respectively.The average recoveries were between 80.1% and 92.5%.
chloramphenicol; tetracycline; solid phase extraction; ultra performance liquid chromatography -tandem mass spectrometry(UPLC-MS/MS)
10.13995/j.cnki.11-1802/ts.011905
学士,工程师(李德刚副教授为通讯作者,E-mail:ldg@sdut.edu.cn)。
2016-05-09,改回日期:2017-02-27
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
中国饲料(2020年7期)2020-06-23
中国猪业(2016年10期)2016-11-16
家庭科学·新健康(2016年5期)2016-05-12
中国药物应用与监测(2015年5期)2015-12-11
四川畜牧兽医(2015年10期)2015-08-15
国外医药(抗生素分册)(2015年3期)2015-07-12
中国药业(2014年21期)2014-05-26
无机化学学报(2014年4期)2014-02-28
