
高通量检测技术对假肥大型肌营养不良分子诊断的研究
2017-09-20 03:16陆静姚如恩朱佳谊王纪文王剑
分子诊断与治疗杂志 2017年5期
陆静 姚如恩★ 朱佳谊 王纪文 王剑
高通量检测技术对假肥大型肌营养不良分子诊断的研究
陆静1姚如恩1★朱佳谊1王纪文2王剑1
目的利用高通量检测技术对假肥大型肌营养不良(DMD/BMD)患者进行分子诊断,检测DMD基因上各种类型的变异,评估组合的分子诊断技术应用于假肥大型肌营养不良患者的价值。方法对106例临床疑似为假肥大型肌营养不良的患者,利用CNVplex®技术检测致病基因拷贝数变异,同时利用靶向捕获及高通量测序检测致病基因单核苷酸水平变异,对患者进行精确的分子诊断。结果通过组合的高通量检测技术,检测到其中85例患者存在DMD基因的致病性变异,包括62例不同大小的外显子缺失和重复,9例单核苷酸水平小缺失和重复,8例无义变异,2例错义变异和2例剪接位点变异。结论分子诊断对假肥大型肌营养不良患者意义重大,组合的高通量检测技术能检测到不同类型的致病基因变异,精确的分子诊断结果对假肥大型肌营养不良患者的诊断和家系的遗传咨询有着重要意义。
高通量测序;分子诊断;基因突变
假肥大型肌营养不良包括Duchenne型肌营养不 良(Duchenne muscular dystrophies,DMD)和Becker型肌营养不良(Becker muscular dystro⁃phies,BMD)。这2种疾病是原发于神经肌肉组织的X连锁隐性遗传的疾病,其共同的分子基础是抗肌萎缩蛋白(dystrophin)编码基因DMD发生不同类型的变异[1]。随着对DMD基因研究的深入,利用分子诊断结果作为DMD/BMD患者的确诊依据,已经越来越得到重视[2]。同时,DMD/BMD患者的分子诊断结果对家系其他相关成员的遗传咨询也有着重要的意义[3]。
作者单位:1.上海交通大学医学院附属上海儿童医学中心,分子诊断实验室,上海200126 2.上海交通大学医学院附属上海儿童医学中心,神经内科,上海200126
DMD基因是人体最大的基因之一,位于X染色体p21.1区域,包含79个外显子,覆盖基因组范围约2 Mb。DMD/BMD患者的变异类型大多为外显子组水平的缺失和重复,占75%左右[4],而单核苷酸水平的变异也可能导致疾病的发生。因此,常规用于单基因遗传病检测的Sanger测序法,通常不作为DMD基因的分子诊断的首选方法。利用多重连接探针扩增技术(multiple liga⁃tion dependent probe amplification,MLPA)检测患者DMD基因外显子水平的拷贝数,被认为是DMD/BMD患者分子诊断的有效手段[5],但在未检测到拷贝数变异的患者中,仍需继续寻找单核苷酸水平的变异。因此,同时检测外显子水平的缺失和重复以及单核苷酸水平的变异是提高DMD/BMD患者分子诊断效率的的最佳手段。本研究拟利用基于MLPA技术改进的外显子拷贝数检测方法CNVplex®,结合靶向基因捕获高通量测序,检测患者DMD基因,探讨分子诊断结果对假肥大型肌营养不良患者的诊断和遗传咨询的意义。
1 材料与方法
1.1 研究对象
收集2014年2月至2016年10月在上海儿童医学中心就诊的疑似假肥大型肌营养不良患者106例,均为男性;采集患者及自愿进行家系验证的患者母亲外周血2 mL EDTA抗凝,4℃保存。使用QIAamp Blood DNA Mini kit核酸抽提试剂盒(Qiagen,德国)从外周血抽提所有样本基因组DNA,经琼脂糖凝胶电泳和紫外分光光度仪检测质量,均符合后续实验要求。所有先证者或监护人已经签署参加本研究的知情同意书。
1.2 CNVplex®检测外显子水平变异
采用天昊生物医药科技(苏州)有限公司针对DMD基因的CNVplex®检测试剂盒。连接反应、连接产物多重荧光PCR扩增、扩增产物荧光毛细管电泳分离均参考试剂盒说明书。PCR扩增产物用美国ABI公司ABI3130XL进行毛细管电泳,原始数据文件采用GeneMapper4.1进行目标峰高的数据读取,进而对样本DMD基因区域的拷贝数进行分析确定,寻找可能存在的外显子水平缺失和重复。
1.3 FastTarget靶向富集及高通量测序
采用天昊生物医药科技(苏州)有限公司设计的FastTarget高通量多重PCR扩增富集技术对DMD基因启动子区、5′非编码区(5′untranslated region,5′UTR)、编码区、转录终止区、内含子剪切点附近8 bp以及有文献报道或数据库记录[6]的已知突变序列进行富集,DNA质量检测、FastTarget目的区域富集、富集产物定量混合、PCR扩增引入接头和特异性标签、样本文库定量混合、文库质量检测均参考试剂盒说明书。富集产物采用高通量测序仪Illumina HiSeq 2000进行测序突变分析,任何一个目的片段的测序深度至少达到20倍。对DMD基因上检测到的致病性单核苷酸变异,设计特异性的PCR扩增引物,Sanger测序验证。
1.4 家系成员检测
在采集到的患者母亲血样的家系中,根据先证者检测到的致病性变异的不同类型,选择特异性的验证方法。若先证者的变异为外显子缺失和重复,则用CNVplex®法对其母亲外周血DNA进行验证。若先证者的变异为单核苷酸水平的变异,则用Sanger测序法进行验证。
2 结果
2.1 106名患者分子诊断结果
在106名患者中,CNVplex®技术检测到62名(51.7%)患者携带有DMD基因上的致病性拷贝数变异,其中包括54个缺失和8个重复,变异的大小从覆盖1个外显子到整个DMD基因不等,对所有的拷贝数变异进行阅读框判断,其中12个为框内突变,50个为框外突变,见图1。通过高通量测序发现21名(17.5%)患者存在单核苷酸水平的变异,包括9例小缺失和重复,8例无义变异,2例错义变异和2例剪接位点变异,具体变异情况见表1,所有变异均通过Sanger测序验证。
2.2 家系成员验证结果
对14例有母亲血样的家系进行致病变异验证后,发现其中6名患者的变异为新发变异,其余8名患者的母亲为与先证者相同变异的杂合携带者。
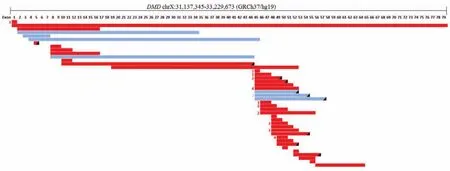
图1 62名患者DMD基因外显子水平的变异统计Figure 1 Eron level variants ofDMDgene in 62 patients

表1 21名检测到DMD基因单核苷酸水平致病性变异Table 1 Single nucleotide level variants ofDMDgene in 21 patients

表2 14个家系验证DMD基因变异结果Table 2 Validation of variants ofDMDgene in 14 pedigrees
3 讨论
假肥大型肌营养不良是常见的累及骨骼肌的神经肌肉疾病,DMD患者病情发展快,3岁左右无法独立行走而需依靠轮椅代步,常死于心肌病或呼吸道并发症,寿命一般不超过20岁[12]。BMD患者病情相对较轻,进展缓慢,主要表现为四肢近端肌肉无力伴腓肠肌肥大。DMD/BMD发病率较高,约1∶5 000[13],对该疾病进行分子诊断,不仅有利于早期准确地诊断疾病,且对家系相关成员的验证将为遗传咨询提供有力的证据[14]。DMD基因的致病性变异影响抗肌萎缩蛋白在横纹肌组织中的表达,导致近端骨骼肌进行性萎缩无力和腓肠肌代偿性肥大。在患者中,DMD基因的外显子缺失最常见,2个常见的热点区域分别位于基因中央区域的第44~53号外显子区域,以及靠近基因5′端的第2~20号外显子区域[15]。除此之外,导致DMD/BMD发病的变异还包括外显子的重复和单核苷酸水平的变异等。
本研究创新地利用基于MLPA技术改良的CNVplex®检测技术,结合FastTarget靶向富集及高通量测序技术,组合应用于检测患者DMD基因外显子水平和单核苷酸水平的变异,试图为患者的分子诊断提供准确的依据。以往的研究认为,MLPA是检测DMD基因外显子水平变异的最佳技术[16],但CNVplex®检测技术相较于 MLPA 在检测探针合成上,更加省时省力。此外通过不同的荧光标记策略,CNVplex®可以同时检测基因组上更多位置的拷贝数信息[17]。
FastTarget技术针对目的区域设计多重PCR扩增体系,对研究者感兴趣的基因组区域捕获富集并进行高通量测序,该技术方法非常成熟,与Sanger测序法相比检测更加快速、操作简单,与其他高通量测序建库方法相比省去了复杂的建库过程[18]。本研究利用这2种检测方法的组合,在106名疑似假肥大型肌营养不良的患者中,成功检测到85个致病性变异,其中外显子水平的变异最小仅覆盖单个外显子,最大覆盖整个DMD基因区域;单核苷酸水平的变异在运用高通量测序方法检出后,均通过Sanger测序法验证。其中9例单核苷酸水平的小缺失和重复以及8例无义变异均会因为终止密码的异常编码而造成截短的蛋白,引起DMD的发生。2例剪接位点变异均未被人类基因突变数据库(Human Gene Mutation Database,HGMD)和 LOVD(Leiden Open Variation Database)数据库收录,但经断裂位点功能预测软件分析(NNSPLICE v9.0)均可能导致异常的RNA剪接。本研究中发现2例错义变异,其中p.Leu116Pro被HGMD 收录[2],而 p.Gln2937Arg为首次发现的错义变异。虽然在之前的DMD/BMD患者相关分子诊断研究中[19],错义变异的检出率相对较低,但Juan⁃Mateu等[20]研究中认为错义变异也可能导致抗肌萎缩蛋白的异常剪接,从而导致疾病的发生,因此必须通过相关功能试验来判断变异是否具有致病性。本研究所采用的组合检测技术在检测成本和报告周期更加符合临床应用的情况下,达到了与以往研究相同水平的检测阳性率(80.2%)[4,21],进一步证实了分子诊断技术在假肥大型肌营养不良患者诊断中的作用。
Monaco等[22]提出的针对DMD基因的“阅读框架假说”(reading frame rule)认为症状较严重的DMD患者大多数是由于框外突变(out of frame)所致,而症状较轻的BMD患者则是由于不影响阅读框的框内突变(in frame)导致。约90%DMD/BMD患者符合阅读框规则,因此基于分子诊断结果判断阅读框,从而确定患者为DMD或BMD,可以在患者出现严重的临床症状前明确诊断,早期干预,提高生活质量[23]。本研究中12例框内突变患者,后期临床均确诊为BMD,与基于阅读框判断的疾病类型相符。患者在检测到DMD基因致病性变异后,应对家系相关成员进行携带者验证,为遗传咨询提供重要线索,本研究发现8名先证者母亲为DMD基因致病变异杂合携带者。在本研究中仍有21名患者并未通过这2种高通量检测技术寻找到DMD基因的致病性变异,可对患者外周血RNA进行逆转录PCR鉴定转录产物是否正常,并进一步寻找内含子中可能存在的致病性变异[4]。此外,也需仔细评估患者临床表型,判断患者是否为肢带型肌营养不良症(limb girdle muscular dystro⁃phy,LGMD)、Emery⁃Dreifuss肌营养不良症(Em⁃ery⁃Dreifuss muscular dystrophy,EDMD)或其他肌营养不良症,并对其进行鉴别诊断[24]。
假肥大型肌营养不良最传统的治疗方法是运用糖皮质激素治疗配合康复理疗。糖皮质激素可以延缓肌力及肌肉功能的衰退,但长时间使用糖皮质激素会出现许多不良反应,并且会出现糖皮质激素耐药情况[25]。基因替代治疗有望成为最有效的DMD/BMD治疗方法,首个批准上市的基因药物deflazacort以外显子跳跃为分子基础,可以成功治疗第51号外显子缺失的患者。德州大学最新研究利用CRISPR⁃Cpf1修复老鼠模型和人类细胞中DMD基因的缺陷,试图达到治疗疾病的目的,为DMD/BMD患者的分子治疗提供了新思路[26]。
准确评估每个患者所携带的DMD基因致病性变异,为其寻找最恰当的分子治疗手段提供了理论依据。
综上所述,利用高通量检测技术,同时检测DMD基因外显子水平的变异和单核苷酸水平的变异,是对DMD/BMD患者进行分子诊断的最佳方法。利用CNVplex®和靶向富集高通量测序技术得到的结果,可以帮助早期诊断为DMD/BMD的患者在一定程度上确诊疾病类型,也为患者寻找可行的个体化治疗方案和对患者家系成员的验证及之后的遗传咨询提供了可靠的依据。
[1]Prior TW,Bridgeman SJ.Experience and strategy for the molecular testing of Duchenne muscular dystrophy[J].J Mol Diagn,2005,7(3):317⁃326.
[2]Flanigan KM,Dunn DM,von Niederhausern A,et al.Mutational spectrum of DMD mutations in dystro⁃phinopathy patients:application of modern diagnostic techniques to a large cohort[J].J Hum Mutat,2009,30(12):1657⁃1666.
[3]Palmucci L,Mongini T,Chiado⁃Piat L,et al.Dystro⁃phinopathy expressing as either cardiomyopathy or Becker dystrophy in the same family[J].J Neurolo⁃gy,2000,54(2):529⁃530.
[4]Takeshima Y,Yagi M,Okizuka Y,et al.Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese re⁃ferral center[J].J Hum Genet,2010,55(6):379⁃388.
[5]Ji X,Zhang J,Xu Y,et al.MLPA application in clini⁃cal diagnosis of DMD/BMD in Shanghai[J].J Clin Lab Anal,2015,29(5):405⁃411.
[6]Aartsma⁃Rus A,van Deutekom JC,Fokkema IF,et al.Entries in the leiden duchenne muscular dystrophy mutation database:an overview of mutation types and paradoxical cases that confirm the reading⁃frame rule[J].J Muscle Nerve,2006,34(2):135⁃144.
[7]Mendell JR,Buzin CH,Feng J,et al.Diagnosis of duchenne dystrophy by enhanced detection of small mutations[J].J Neurology,2001,57(4):645⁃650.
[8]Prior TW,Wenger GD,Papp AC,et al.Rapid DNA haplotyping using a multiplex heteroduplex approach:application to Duchenne muscular dystrophy carrier testing[J].J Hum Mutat,1995,5(3):263⁃268.
[9]Deburgrave N,Daoud F,Llense S,et al.Protein⁃and mRNA ⁃based phenotype⁃genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene[J].J Hum Mutat,2007,28(2):183⁃195.
[10]van Essen AJ,Busch HF,te Meerman GJ,et al.Birth and population prevalence of Duchenne muscular dys⁃trophy in the Netherlands[J].Hum Genet,1992,88(3):258⁃266.
[11]Roest PA,van der Tuijn AC,Ginjaar HB,et al.Ap⁃plication of in vitro myo⁃differentiation of non⁃muscle cells to enhance gene expression and facilitate analysis of muscle proteins[J].Neuromuscul Disord,1996,6(3):195⁃202.
[12]Hermans MC,Pinto YM,Merkies IS,et al.Heredi⁃tary muscular dystrophies and the heart[J].Neuromus⁃cul Disord,2010,20:479⁃492.
[13]Dooley J,Gordon KE,Dodds L,et al.Duchenne muscular dystrophy:a 30⁃year population⁃based inci⁃dence study[J].Clin Pediatr(Phila),2010,49(2):177⁃179.
[14]中华医学会神经病学分会.中国假肥大型肌营养不良症诊治指南[J].中华神经科杂志,2016,49(1):17⁃20.
[15]Oudet C,Hanauer A,Clemens P,et al.Two hot spots of recombination in the DMD gene correlate with the deletion prone regions[J].Hum Mol Genet,1992,1(8):599⁃603.
[16]Yang J,Li SY,Li YQ,et al.MLPA⁃based genotype⁃phenotype analysis in 1053 Chinese patients with DMD/BMD[J].BMC Med Genet,2013,14:29.
[17]Zhang X,Xu Y,Liu D,et al.A modified multiplex li⁃gation⁃dependent probe amplification method for the detection of 22q11.2 copy number variations in pa⁃tients with congenital heart disease[J].BMC Genom⁃ics,2015,16:364.
[18]Jiang T,Tan L,Chen Q,et al.A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese[J].Neurobiol Aging,2016,42:217.e1⁃3.
[19]白莹,李双,宗亚楠,等.杜氏/贝氏肌营养不良症433个家系的基因突变分析[J].中华医学杂志,2016,96(16):1261⁃1269.
[20]Juan⁃Mateu J,Gonzalez⁃Quereda L,Rodriguez MJ,et al.DMD mutations in 576 dystrophinopathy fami⁃lies:a step forward in genotype⁃phenotype correlations[J].Plos One,2015,10(8):e0135189.
[21]Hofstra RM,Mulder IM,Vossen R,et al.DGGE⁃based whole⁃gene mutation scanning of the dystrophin gene in duchenne and becker muscular dystrophy pa⁃tients[J].Hum Mutat,2004,23(1):57⁃66.
[22]Monaco AP,Bertelson CJ,Liechti⁃Gallati S,et al.An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus[J].Genomics,1988,2(1):90⁃95.
[23]Kesari A,Pirra LN,Bremadesam L,et al.Intergrated DNA,cDNA,and protein studies in becker muscular dystrophy show high exception to the reading frame rule[J].Hum Mutat,2008,29(5):728⁃737.
[24]Schwartz M,Hertz JM,Sveen ML,et al.LGMD2I presenting with a characteristic duchenne or becker muscular dystrophy phenotype[J].Neurology,2005,64(9):1635⁃1637.
[25]许田田,蓝丹.假性肥大型肌营养不良的治疗进展[J].中国当代儿科杂志,2015,17(3):294⁃298.
[26]Zhang Y,Long C,Li H,et al.CRISPR⁃cpf1 correc⁃tion of muscular dystrophy mutations in human cardio⁃myocytes and mice[J].Sci Adv,2017,3(4):e1602814.
Analysis of molecular diagnosis in DMD/BMD patients using high throughput detection technique
LU Jing1,YAO Ruen1★,ZHU Jiayi1,WANG Jiwen2,WANG Jian1
(1.Molecular Diagnostic Laboratory,Shanghai Children's Medical Center Affiliated to Shanghai Jiaotong University School of Medicine,Shanghai,China,200126;2.Department of Neurology,Shanghai Children's Medical Center Affiliated to Shanghai Jiaotong University School of Medicine,Shanghai,China,200126)
ObjectiveTo perform molecular diagnosis for patients suspected with dystrophinopathy,and to detect pathogenic variants onDMDgene with the aim of evaluation of molecular diagnosis in those patients.MethodCNVplex®technology and targeted capture combined with high throughput sequencing were used to detect variants of exon level and single nucleotide level respectively in 106 suspected dystrophinopathy patients.Results85 pathogenic variants were detected through combined detection,including 62 exon deletion/duplication,9 nucleotide insertion/deletion,8 nonsense variants,2 missense variants and 2 splicing variants.ConclusionMolecular diagnosis is important for patients suspected with dystrophinopathy and combined testing strategy manifested better efficiency.The accurate results of molecular diagnosis offers great help in both clinical diagnosis and family genetic counseling.
High throughput sequencing;Molecular diagnosis;Genetic variants
★通讯作者:姚如恩,E⁃mail:yaoruen@126.com
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
中国生殖健康(2020年4期)2021-01-18
透析与人工器官(2020年1期)2020-11-16
铁道通信信号(2019年8期)2019-10-10
中国生殖健康(2018年4期)2018-11-06
中国发展观察(2017年8期)2017-04-26
中国当代医药(2015年33期)2015-03-01
