
第一性原理对CdnSen (11≤n≤16)和Cd15Se16Ag纳米团簇电子性质研究
2017-09-15 07:17索凤怡
哈尔滨商业大学学报(自然科学版) 2017年4期
索 凤 怡
(天津工业大学 理学院,天津 300387)
第一性原理对CdnSen(11≤n≤16)和Cd15Se16Ag纳米团簇电子性质研究
索 凤 怡
(天津工业大学 理学院,天津 300387)
由于自身电子性质的可协调性,镉硫族半导体尤其是掺杂的纳米半导体团簇吸引了广泛关注.采用密度泛函理论的方法对CdnSen(11≤n≤16) 和 Cd15Se16Ag稳定结构进行了模拟研究分析.考虑到Ag原子掺杂在Cd16Se16纳米团簇中位置的不同,主要讨论了两种可能的掺杂位(S1, S2).基于此,详细地分析了Cd15Se16AgS1和 Cd15Se16AgS2团簇的几何结构和电子性质.Ag原子的掺杂改变了其自身的对称群和大小,而Ag原子掺杂位置的不同也被重点关注.此外,详细分析了结合能、带隙、态密度和电荷密度分布.结果表明掺杂后引入了一个新的能级,该能级被称为受主能级.介于价带和导带间的受主能级电荷密度的贡献主要来自于Se原子的p轨道和Ag原子的d轨道.这些说明了Ag原子掺杂和掺杂位置是两个影响着团簇电子性质的重要原因.
CdSe;Ag掺杂;密度泛函理论;结构和电子性质
作为一种典型的Ⅱ-Ⅵ族半导体材料,CdSe纳米团簇已经引起了半导体科学工作者的极大关注,是当今研究的热点之一.由于量子限域效应的影响[1-2],通过改变尺寸大小,CdSe纳米团簇的几何结构和电学性质可以因此发生改变,这和块状材料的性质是不同的.在实验中,CdSe材料被广泛的应用于太阳能电池[3]、中红外光电探测器[4]、发光二极管等光电纳米器件中[5].理论上为了进一步研究它的性质,我们可以通过第一性原理的方法计算和分析CdnSen纳米团簇的性质.由此,在Materials Stutio这个软件中,通过改变n值的大小,不同尺寸大小的CdnSen纳米团簇结构被构造出来[6-9].分析结果表明,当n的大小发生改变时,CdnSen纳米团簇的几何对称性、结合能、带隙、状态密度函数以及电荷密度分布都会因此而改变.J·Wang[10]等人发现了当n>10时,CdTe纳米团簇有两种同分异构体,一种是内嵌式(壳核)结构,另外一种是空的笼状团簇结构.
掺杂也是一种可以改变纳米团簇自身几何结构和电学性质的方法[11].通过改变掺杂原子,原有的CdnSen纳米团簇将变成p型或者N型半导体.S·K·Bhattacharya和A Kshirsagar研究证明了当CdTe团簇中的一个Cd原子被Pb/Ag原子替换掉时整体团簇将成为典型的P型半导体,而当In原子替换掉其中的一个Cd原子时将变成典型的N型半导体[12].此外,S Xu等人研究表明由于Ag原子的掺杂,原有的ZnSe团簇结构将变得更稳定[13].随着对Ⅱ-Ⅵ族半导体量子点研究的不断深入,CdSe团簇的研究也越来越重要.然而,对于中等大小的CdnSen以及Ag原子掺杂的CdnSen团簇的研究未见报道.
本文采用密度泛函理论第一性原理的方法建模并分析了中等尺寸大小的CdnSen(11≤n≤16)纳米团簇的几何结构和电学性质.通过比较结合能大小,可以发现Cd16Se16具有较高的结构对称性和稳定性.因此,在Cd16Se16空的笼状纳米团簇中,我们用Ag原子替换掉其中的一个Cd原子并研究其性质的改变.由于掺杂原子的位置不同,其掺杂后团簇的稳定性和电学性质都会因此而发生改变.结构对称性、结合能、带隙、电荷密度分布以及最高占据轨道和最低为占据轨道这些性质都将逐一的分析和讨论.
1 计算方法
本文使用密度泛函理论[14-15]第一性原理的方法对CdnSen(11≤n≤16)以及Cd15Se16Ag团簇的结构和电学性质进行理论计算和分析.鉴于CASTEP模块耗时大的缺陷,我们采用Materials Stutio中DMOL3模块去进行分析和计算[16].在交换关联势的选取上,由于我们研究的团簇尺寸较大,所以我们不采用局域密度近似(LDA),而选择分子系统中更为可靠的广义梯度近似(GGA)[17],GGA的具体形式选择的是Perdew,Burke和Ernzerhof三人提出的PBE近似[18].
另外,计算参数的选择是非常重要的一个环节,选择的不同直接导致的结果的精确与否.DMOL3模块下有四个精度可以选择,分别为Coarse、Medium、Fine、Customized,在优化和分析结构时都选用的精度最高的一级.在优化参数的设定上,选择几何优化的同时,不需要选择自旋极化和对称约束.另外,我们选择DNP基组计算,所对应的自洽允许误差(SCF)设定为10-6Ha,这足以保证了计算的精度.最大的SCF周期和轨道截断能分别设定为1 000和4.6Å.基于此,去计算它的频率,福井函数和布居分析.优化结束后,可以得到它最稳定的基态结构,由此我们可以去计算其结合能的大小.为了去分析它的状态密度分布(DOS)和能带结构图,需要去建立一个大斜方晶系的超晶胞.将CdnSen团簇放置于超晶胞的中心,团簇表面原子距离晶胞边缘大于20 Å,这足以避免了团簇与团簇之间的相互作用.
在引入掺杂原子(Ag原子)时,由于Ag原子取代了一个原有的Cd原子,导致原来的Cd16Se16团簇失去一个电子,这时我们应该采用自旋极化的方式进行结构优化.在分析能量和态密度时,其他的参数和掺杂前的参数设置保持一致.
2 结果与分析
2.1 CdnSen(11≤n≤16)团簇结构及电子性质分析
在对CdnSen(11≤n≤16)半导体团簇的初始构型进行几何优化后,图1给出了其最低能量结构.从图中可以看出,CdnSen(11≤n≤16)团簇是由Cd原子和Se原子组成的四元环和六元环交替构成.其中,一个四元环包含了2个Cd原子和2个Se原子而一个六元环包含了3个Cd原子和3个Se原子.其中,Cd11Se11团簇中Cd原子和Se原子之间的键长约为2.57Å-2.59 Å而Cd13Se13、Cd14Se14、Cd15Se15、Cd16Se16团簇中的Cd原子和Se原子间键长分别为2.54Å-2.58Å、2.57Å-2.59Å、2.57Å-2.60Å、2.60Å-2.80Å.值得注意的是,在Cd12Se12团簇中,Cd原子和Se原子间键长都为2.58 Å.由于n值的变化,团簇的对称性也会相应发生改变.从图1可以看出,Cd11Se11、Cd12Se12、Cd13Se13、Cd14Se14、Cd15Se15和Cd16Se16团簇的对称群分别为Cs、Th、C3v、Cs、C3h和Td.其中,由于Th和Td属于立方晶系,所以Cd12Se12和Cd16Se16团簇具有较高的结构对称性.通过测量团簇中最远两个原子之间的距离,我们可以知道团簇的大小.表1给出了CdnSen(11≤n≤16)纳米团簇大小.从表1中可知,Cd12Se12团簇的大小较之其他五个最小,约为8.48 Å.
其中,浅棕色球代表Cd原子,深棕色球代表Te原子.
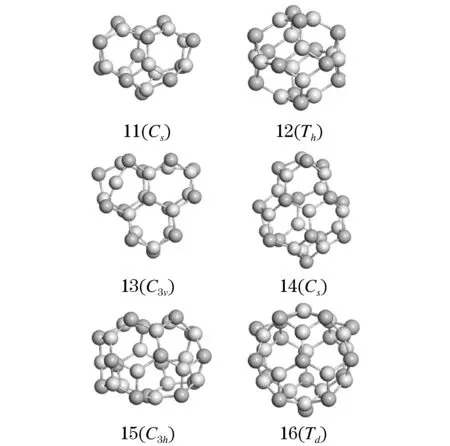
图1 CdnSen (11≤n≤16)纳米团簇的几何结构

StructureCd11Se11Cd12Se12Cd13Se13Cd15Se15Cd16Se16L(Å)9.248.4810.309.959.64
为了去比较团簇的稳定性,我们可以通过计算其结合能的大小.对于未掺杂的团簇来说,其平均结合能的公式为:
Eb(n)={[n×E(Cd)+n×E(Se)]-E[(CdSe)n]}/n
其中:代表着所有单Cd原子的能量,代表着所有单Se原子的能量,E((CdSe)n)代表着CdnSen团簇的能量.通过计算可以得到CdnSen团簇的平均结合能.图2给出了CdnSen(11≤n≤16)的平均结合能.从图中可以看出,当n>13时结合能在不断变大,且n<13时结合能也是在变大.这就意味着n=13时团簇的结合能最小.也就是说对于11≤n≤16的CdSe团簇来说,Cd13Se13团簇稳定性最低.相反的,Cd16Se16团簇的结合能为3.93 eV,在这些团簇中稳定性最高.

图2 CdnSen (11≤n≤16)的平均结合能
最低未占据原子轨道(LUMO)和最高占据原子轨道(HOMO)的差值即带能带隙的大小在图3中给出.通过比较带隙的值,我们可以知道电子想要吸收光子发生跃迁从而导电的难易程度.从图3可以看出,Cd11Se11、Cd12Se12、Cd13Se13、Cd14Se14、Cd15Se15和Cd16Se16团簇的带隙分别为2.08、2.07、1.95、2.03、2.07、2.15 eV.其中,Cd16Se16的带隙最大,也就是说团簇需要吸收更多的能量使得电子从低的未占据轨道跃迁到高的占据轨道.由于尺寸大小的改变,带隙相应的也发生了很大的改变,这也证实了半导体纳米团簇的带隙和自身的大小有着密切的联系[1].
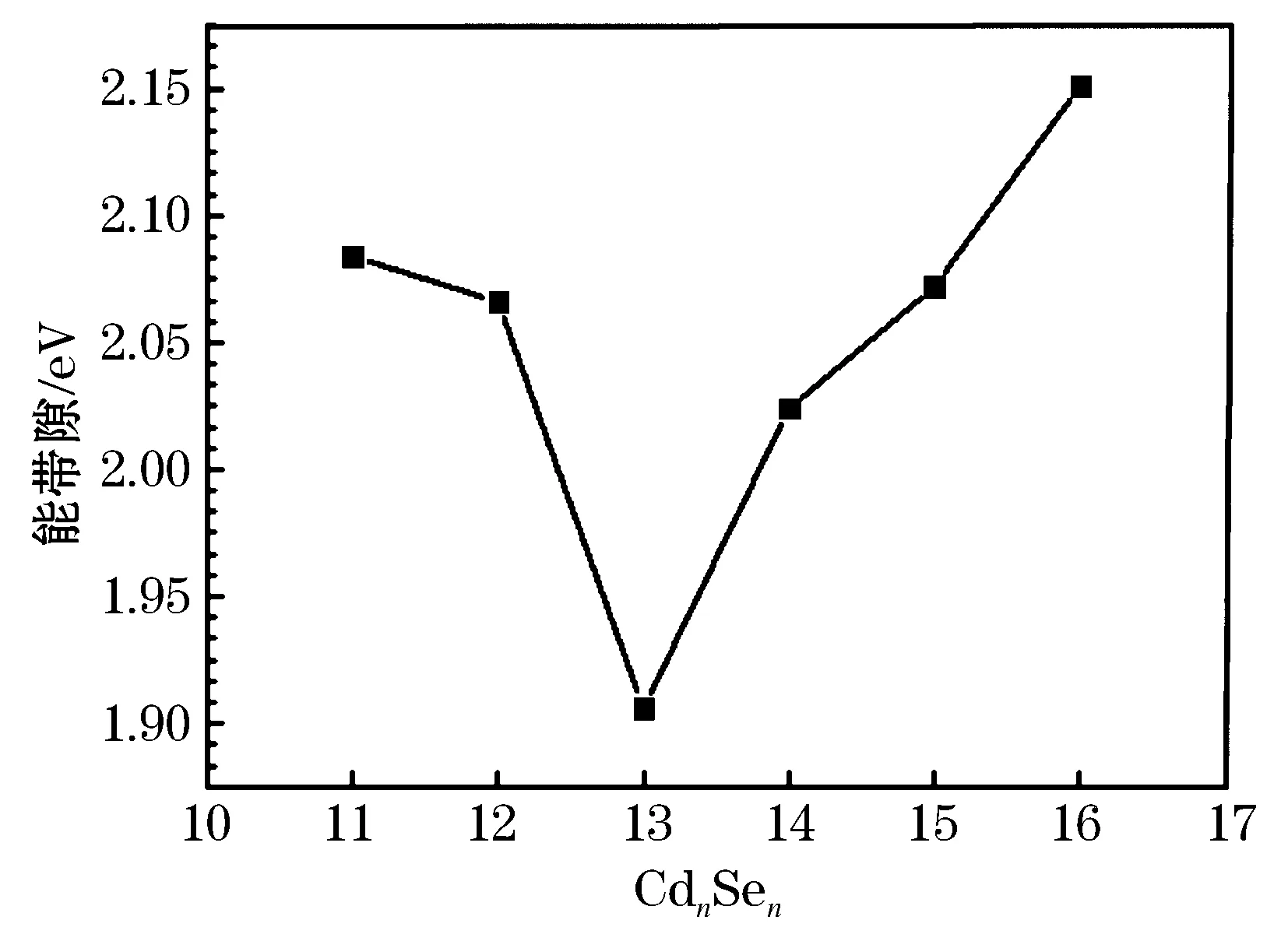
图3 CdnSen (11≤n≤16) 团簇的LUMO-HOMO的能带隙
2.2 Cd15Se16Ag团簇结构及电子性质分析
鉴于掺杂可以改变半导体原有的几何结构和电子性质[12],我们引入一个原子(Ag原子)来替换掉原有团簇中的一个Cd原子.对于CdnSen(11≤n≤16)纳米团簇来说,Cd16Se16团簇具有高稳定性和良好的几何对称性,因此我们选择替换Cd16Se16团簇中的一个Cd原子.考虑到掺杂位置的不同,图4给出了两种不同的掺杂位所对应的Cd15Se16Ag团簇优化好的结构图.其中,S1代表着Ag原子取代了两个六元环和一个四元环所夹着的Cd原子而S2代表着Ag原子取代了三个六元环所夹着的Cd原子.通过比较对称群,我们发现Cd15Se16AgS1属于Cs对阵而 Cd15Se16AgS2属于C3v对称.而未掺杂的Cd16Se16团簇属于Td对称.这就说明了掺杂改变了团簇原有的几何对称性.此外,从表2可以看出团簇的大小较之未掺杂前略微有点变化.
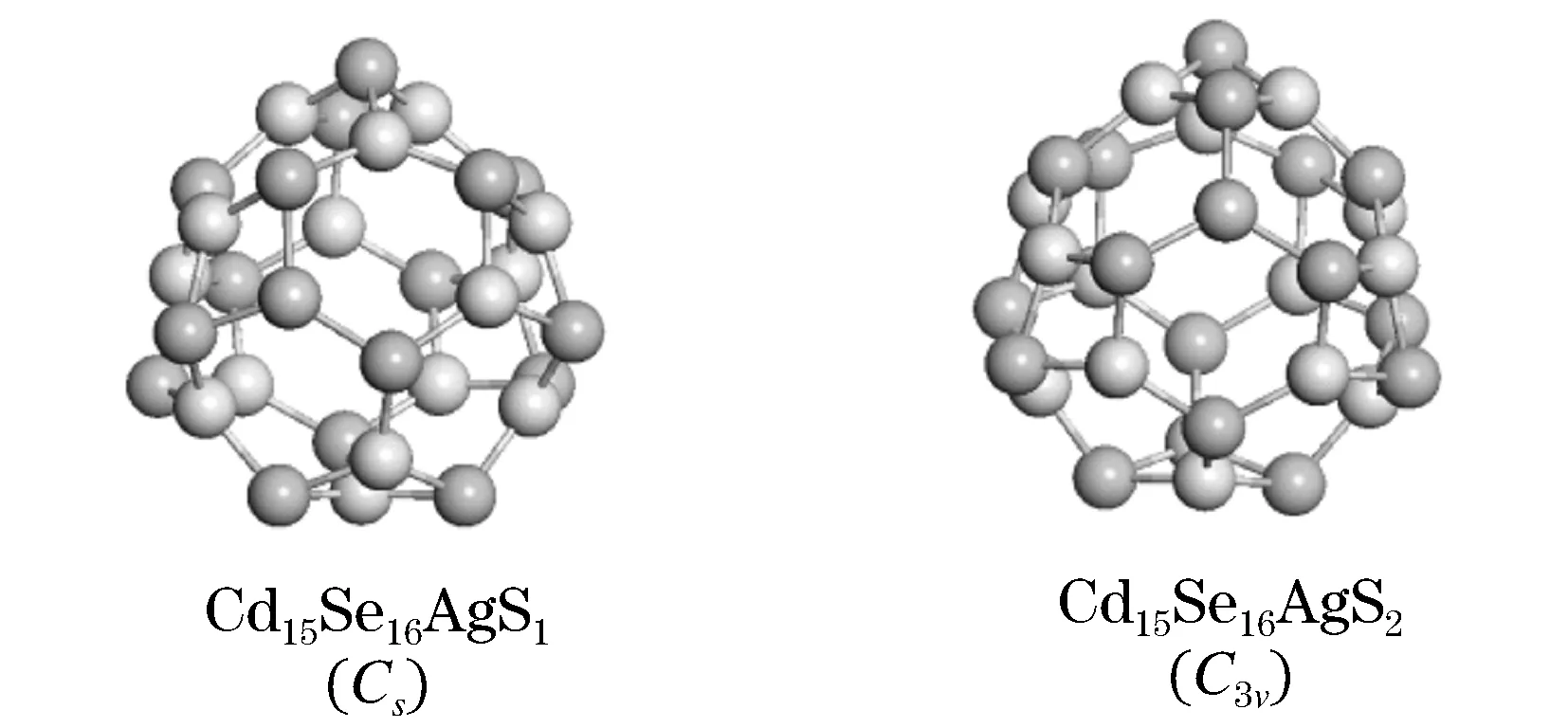
图4 Cd15Se16AgS1 和 Cd15Se16AgS2纳米团簇的几何结构
其中:浅棕色球表示Cd原子,深棕色球表示Te原子,蓝色球表示Ag原子.
表2Cd15Se16AgS1和Cd15Se16AgS2纳米团簇的大小L(Å),整体的结合能Eb和HOMO-LUMO带隙
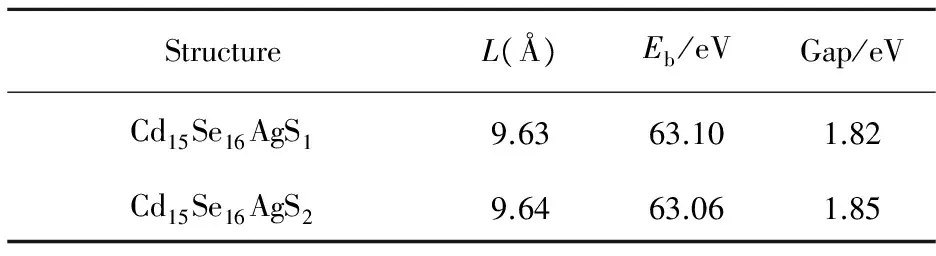
StructureL(Å)Eb/eVGap/eVCd15Se16AgS19.6363.101.82Cd15Se16AgS29.6463.061.85
掺杂后为了比较结合能的改变,我们可以通过计算Cd15Se16Ag团簇整体的结合能,其公式如下:
Eb=[15×E(Cd)+16×E(Se)+E(Ag)]-E(Cd15Se16Ag)
其中:E(Cd)代表着所有单Cd原子的能量,E(Se)代表着所有单Se原子的能量,E(Cd15Se16Ag)代表
着Ag原子的能量,代表着Cd15Se16Ag团簇的能量.从表2可以看出,Cd15Se16AgS1和 Cd15Se16AgS2团簇的结合能为63.10 eV和63.06 eV,然而Cd16Se16团簇的整体结合能为62.80 eV,由此可见,掺杂后的团簇结合能要比掺杂前大,即掺杂Ag原子提高了其自身的稳定,这也证明了金属原子掺杂会提高半导体团簇的稳定性[13].此外,Cd15Se16AgS1和 Cd15Se16AgS2团簇的结合能也略有不同,这说明掺杂位置的不同会对结合能造成影响.
此外,表2还给出了掺杂后的带隙,Cd15Se16AgS1和 Cd15Se16AgS2团簇的HOMO-LUMO带隙分别为1.82 eV和1.85 eV,而未掺杂的Cd16Se16团簇的带隙为2.15 eV.由此可见,由于Ag原子的掺杂,原有团簇的带隙发生了很大改变,即电子从低的未占据轨道跃迁到高的占据轨道更容易.Ag原子在不同的掺杂位置对带隙会有一定程度的影响,但影响不大.
为了进一步研究能级的分布和不同轨道对能级的贡献,画出了Cd16Se16、Cd15Se16AgS1和 Cd15Se16AgS2团簇的整体态密度图和局部态密度图,其中费米能级移动到0 eV.从图5可以看出,能带分为了两个部分,在费米能级左侧称之为价带而在费米能级右侧称之为导带.当掺杂Ag原子后,由于团簇整体失去了一个垫子,此时团簇变成了一个典型的p型半导体,Ag原子所处的能级称之为守住能级,如图中的Ea所示.通过比较可以看出,相比于Cd15Se16AgS2,Cd15Se16AgS1团簇的Ea更靠近价带底.PDOS图给出了s轨道、p轨道以及d轨道对每个能级的贡献.其中,Ea所处位置的能级贡献主要来自于p轨道和少量的d轨道的贡献,价带顶附近主要来自于p轨道和极少部分d轨道的贡献而导带底附近主要来自于s轨道和p轨道的贡献.
图6给出了Cd16Se16和Cd15Se16AgS1团簇的HOMO和LUMO的电荷密度分布图. 其中,费米位置移动到0 eV.从图6中可以看出,由于Ag原子掺杂的影响,对于原来的Cd16Se16的HOMO和LUMO分布造成很大的影响.Cd15Se16AgS1团簇的HOMO分布主要聚集在Ag原子的周围,其中Ag原子d轨道的贡献显而易见.然而对于Cd15Se16AgS1的LUMO的影响则为电荷主要分布于远离Ag原子的一侧.
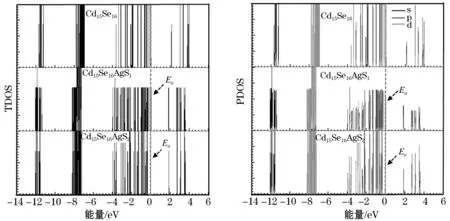
图5 Cd15Se16AgS1 和 Cd15Se16AgS2团簇的整体态密度和局部态密度图
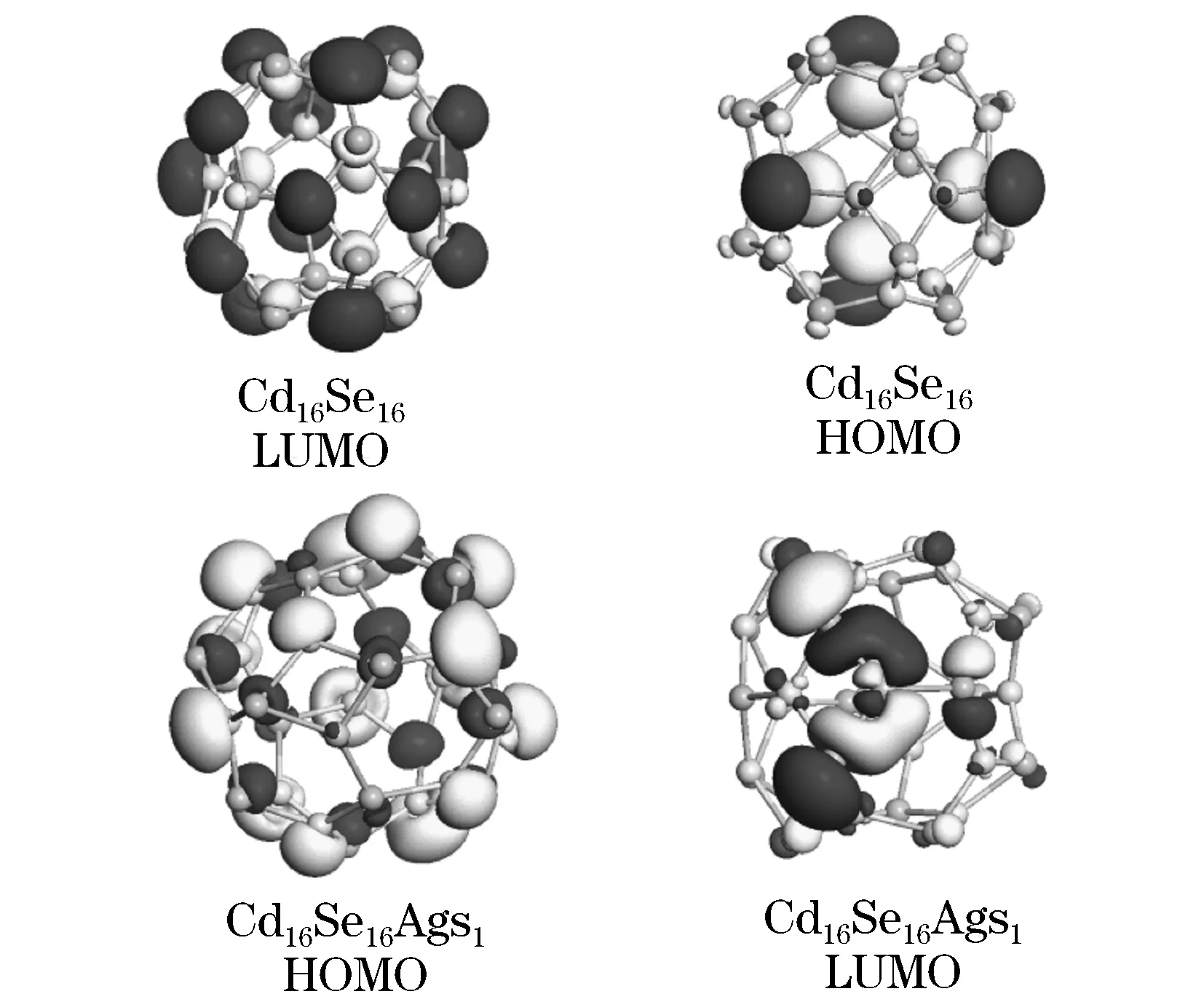
图6 Cd15Se16AgS1 和 Cd15Se16AgS2团簇的部分电荷密度
3 结 语
本文基于密度泛函理论第一性原理的方法对CdnSen(11≤n≤16)团簇结构及电子性质进行了几何优化和电子性质的模拟分析.由于n的不同,几何结构和对称群也会发生相应的改变.通过比较平均结合能,结果显示当n=16时团簇具有高稳定性,所以用Ag原子掺杂在Cd16Se16团簇中研究其电子性质的变化.考虑到掺杂位置的不同,我们将详尽地讨论Cd15Se16AgS1和 Cd15Se16AgS2这两种团簇性质的不同.通过比较结合能发现,掺杂后团簇的结合能较之掺杂前大,也就意味着Ag原子的掺杂提高了Cd16Se16团簇的稳定性,而由于掺杂位置的不同,结合能的大小也略微有所改变.由于Ag原子的掺杂,引入了一个新的能级,即受主能级Ea,而对于Cd15Se16AgS2来讲,Cd15Se16AgS1团簇的Ea更靠近于价带底.通过电荷密度分布图,可以明显看出Ag原子对原有的HOMO和LUMO的电荷分布将造成很大的影响.
[1] NORRIS D J, BAWENDI M G. Measurement and assignment of the size-dependent optical spectrum in CdSe quantum dots [J]. Phys Rev B Condens Matter, 1996, 53(24): 16338-16346.
[2] PENG X, MANNA L, YANG W,etal. Shape control of CdSe nanocrystals [J]. Nature, 2000, 6773(404): 59-61.
[3] JIAO S, SHEN Q, MORASERO I,etal. Band engineering in core/shell ZnTe/CdSe for photovoltage and efficiency enhancement in exciplex quantum dot sensitized solar cells [J]. Acs Nano, 2015, 9(1): 908-915.
[4] JEONG K S, SIONNEST P G. Mid-infrared photoluminescence of CdS and CdSe colloidal quantum dots [J]. Acs Nano, 2016, 10, 2225-2231.
[5] KIM B H, ONSED M S, LIM J B,etal. High-resolution patterns of quantum dots formed by electrohydrodynamic jet printing for light-emitting diodes [J]. Nano. Lett, 2015, 15(2): 969-973.
[6] 刘朝霞, 楚合营, 胡云莎, 等. CdnSen(1≤n≤12)团簇结构与电子性质的第一性原理研究 [J]. 2011, 28(6): 1037-1044.
[7] GUTSEV L G, DALAL N S, RAMACHANDRAN B R,etal. Spectral signatures of semiconductor clusters: (CdSe)16 isomers [J]. Chem. Phys. Lett, 2015, 636: 121-128.
[8] PUZDER A, WILLIAMSON A J, GYGI F,etal. Self-healing of CdSe nanocrystals: first-principles calculations [J]. Phys. Rev. Lett, 2004, 92(21): 217401.
[9] JOSE R, ZHANPEISOV N U, FUKUMURA H,etal. Structure-property correlation of CdSe clusters using experimental results and first-principles DFT calculations [J]. J. Am. Chem. Soc, 2006, 128(2): 629-636.
[10] WANG J, MA L, ZHAO J,etal. Structural growth behavior and polarizability of CdnTen(n=1-14) clusters [J]. J. Chem. Phys, 2009, 130(21): 8706.
[11] LAMIA N, ESTEVES R J A, SHOPAN H,etal. Metal semiconductor hybrid aerogels: evolution of optoelectronic properties in a low-dimensional CdSe/Ag nanoparticle assembly [J]. Acs .nano, 2015, 9(10): 9810.
[12] BHATTACHARYA S K, KSHIRSAGAR A. Defect studies in small CdTe clusters [J]. Eur. Phys. J. D, 2011, 61(3): 609-619.
[13] XU S, WANG C, WANG Z,etal. Theoretical and experimental investigation of stabilityand spectra of doped Ag: ZnSe nanocrystals [J]. J. Mol. Model, 2014, 20(4): 2184.
[14] CAR R, PARRINELLO M. Unified approach for molecular dynamics and density-functional theory [J]. Phys. Rev. Lett, 1985, 55(22): 2471.
[15] JONES R O. Density functional theory: Its origins, rise to prominence, and future [J]. Review of Modern Physics, 2015, 87(3): 897-923.
[16] DELLEY B. From molecules to solids with the DMol3approach [J]. J. Chem. Phys, 2000, 113(18): 7756-7764.
[17] PERDEW J P, BURKE K, WANG Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system [J]. 1998, Phys. Rev. B, 54: 16533.
[18] HAMMER B, HANSEN L B, NORSKOV J K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals [J]. Phys. Rev. B, 1999, 59: 7413.
StudyonelectricalinvestigationofCdnSen(11≤n≤16)andCd15Se16Agnanoclustersbyfirstprincipletheory
SUO Feng-yi
(School of Science, Tianjin Polytechnic University, Tianjin 300387, China)
Cadmium chalcogenide semiconductor, especially doped nanoclusters, have attracted so much concern for their tunability of its electronic properties. The ultra-stable CdnSen(11≤n≤16) and Cd15Se16Ag structures were investigated by density functional theory. Considering the effect of doping site in Ag-doped Cd16Se16nanocluster, two possible substitutional sites (S1, S2) were discussed. Based on it, the structural and electrical properties of Cd15Se16AgS1and Cd15Se16AgS2were studied in detail. Indeed, the Ag atom doping affected its original symmetry group and size. Particular attention was paid to the effects of Ag substitutional sites. Furthermore, the bingdingenergy, energy gap, density of states and charge density distributions were analyzed, revealing that one new energy level called acceptor level was introduced in the original system. The charge distribution of acceptor level which was between valence band and conduction band, mainly from p-orbital of Se atoms and d-orbital of Ag atom. These illustrate that metal (Ag atom) doping and substitutional sites are two important factors for its electrical properties.
CdSe; Ag doping; density functional theory; structural and electrical properties
2016-10-12.
索凤怡(1992-),女,硕士,研究方向:半导体纳米团簇.
TB383
:A
1672-0946(2017)04-0444-06
猜你喜欢
大学物理(2022年9期)2022-09-28
物理通报(2020年7期)2020-07-01
复旦学报(医学版)(2020年3期)2020-06-18
原子与分子物理学报(2020年5期)2020-03-17
制冷(2019年2期)2019-12-09
华人时刊(2019年15期)2019-11-26
中学生数理化·高三版(2017年1期)2017-04-20
中国惯性技术学报(2015年1期)2015-12-19
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
原子与分子物理学报(2015年3期)2015-11-24
