
新生儿筛查发现4例MCCD患儿的临床和基因分析*
2017-09-11 09:39王彦云孙云程威蒋涛
临床检验杂志 2017年8期
王彦云,孙云,程威,蒋涛
(南京医科大学附属妇产医院遗传医学中心,南京 210004)
·临床实验研究·
新生儿筛查发现4例MCCD患儿的临床和基因分析*
王彦云,孙云,程威,蒋涛
(南京医科大学附属妇产医院遗传医学中心,南京 210004)
目的 对4例经由新生儿筛查发现的不同临床类型的3-甲基巴豆酰辅酶A羧化酶缺乏症(3-methylcrotonyl-coenzyme A carboxylase deficiency,MCCD)患儿,用尿气相色谱质谱和基因分析证实其诊断。方法 对新生儿筛查中C4DC+C5OH>0.6 μmol/L的新生儿召回复查,同时检测其母亲的C4DC+C5OH浓度。用尿气相色谱质谱分析对MCCD疑诊病例进行临床诊断,再通过基因分析进一步证实。结果 通过基因诊断确诊MCCD 3例,包括MCCD患儿1例,父源性MCCD 1例,母源性MCCD 1例。另有1例临床诊断的MCCD患儿经基因检测仅找到1个致病位点。结论 对新生儿筛查中发现的C4DC+C5OH增高的新生儿家系(包括母亲和父亲)应行MS/MS检测。疑似MCCD患者基因检测仅发现1个致病位点时不要轻易否定MCCD的诊断,建议定期随访。
3-甲基巴豆酰辅酶A羧化酶缺乏症;串联质谱;3-羟基异戊酰肉碱;母源性;杂合子
3-甲基巴豆酰辅酶A羧化酶缺乏症(3-methylcrotonyl-coenzyme A carboxylase deficiency,MCCD)作为一种亮氨酸代谢障碍所致的常染色体隐性遗传的有机酸代谢缺陷病,在1970年由Eldjarn等[1]首次报道。其致病基因为MCCC1和MCCC2,致病基因的突变可以导致亮氨酸代谢途径中的3-甲基巴豆酰辅酶A羧化酶(3-methylcrotonyl-coenzyme A carboxylase,MCC)缺乏。目前MCCD在欧美国家某些最早开展串联质谱(Tandem mass spectrometry,MS/MS)技术的地区发病率约为1/36 000[2]。我院遗传医学中心于2013年开始建立MS/MS技术检测平台并用于新生儿筛查,对于新生儿筛查中C4DC+C5OH超过0.6 μmol/L的新生儿召回复查,并常规对其母亲进行MS/MS检测分析,已经确诊新生儿MCCD 1例、母源性MCCD 1例、父源性MCCD 1例和1例仅找到1个杂合突变的疑似患儿。针对母源性MCCD和杂合突变患者的研究较为罕见。本研究将新生儿筛查中发现的MCCD患儿的筛查指标,临床表现以及基因突变分析结果综合分析,以期提高临床工作者对此疾病的临床认识和诊疗水平。
1 资料与方法
1.1 研究对象 2013年12月至2016年11月本实验室用MS/MS技术对65 451例出生3~5 d的新生儿进行遗传代谢病筛查,筛查样本的采集严格按照新生儿采血规范执行。对于初次筛查结果中C4DC+C5OH浓度>0.6 μmol/L的新生儿召回复查,并对其母亲进行MS/MS检测分析。
1.2 其他有机酸代谢病的鉴别诊断 采用尿气相色谱质谱(Gas chromatography-mass spectrometry,GC/MS)检测技术(杭州博圣公司提供)对其他可导致C4DC+C5OH增高相关的有机酸代谢病(包括多种酰基CoA羧化酶缺乏症、3-甲基戊二酰CoA水解酶缺乏症、3-羟-3甲基戊二酰CoA裂解酶缺乏症和β-酮硫解酶缺乏症)进行鉴别诊断,均由本实验室完成。结果判读:MCCD患儿尿片中可检测到特异性的3-甲基巴豆酰甘氨酸(3- methylcrotonyl -glycine,3-MCG)和3-羟基异戊酸(3-hydroxy-isovalerate,3-HIVA)。
1.3 基因诊断 在知情同意的原则下,采集患儿及其父母外周血,EDTA-Na2抗凝,置-20 ℃冻存。采用基因组DNA提取试剂盒(美国Omega公司)提取患儿及其父母外周静脉血的基因组DNA。
1.3.1 目的基因PCR扩增 按照Ion AmpliSeqTMInherited Disease Panel 试剂盒(美国Life technologies公司)说明书操作对MCCD的致病基因MCCC1和MCCC2基因的所有外显子进行扩增, PCR扩增反应体系为20 μL,包括10 ng基因组DNA,1×Ion AmpliSeqTMHiFi Master Mix和1×Ion AmpliSeqTMPrimer Pool,ddH2O补足体积。循环参数:9 ℃酶激活2 min;99 ℃变性15 s,60 ℃退火及延伸8 min,共12个循环;10 ℃保存。PCR反应完毕后加2 μL FuPa ReagentTM进行引物消化,以去除部分引物。反应条件:50 ℃ 10 min;55 ℃ 10 min, 65 ℃ 20 min。采用Ion XpressTMBarcode Adapters 1-16 Kit(批号:4471250,美国Life Technologies公司) 对样品加上序列标签及Ion Torrent个体化基因组测序仪(personal genome machine,PGM)测序平台兼容的测序接头。
1.3.2 文库构建 采用Ion AmpliSeqTMLibrary Kit 2.0(批号:4475345,美国Life Technologies公司)进行文库构建。制备的文库平均片段大小约为300 bp。
1.3.3 乳液PCR和ISPs富集 采用Ion PGM 200 Xpress Template Kit(批号:4474280,美国Life Technologies公司)在Ion OneTouch仪(美国Life Technologies公司)上进行乳液PCR,挂有模板的磁珠颗粒(Ion Sphere Particles,ISPs)富集在Ion OneTouch ES仪(美国Life Technologies公司)上完成。
1.3.4 Ion Torrent PGM平台测序 采用Ion PGM 200 Sequencing Kit(批号:4474004,美国Life Technologies公司)及Ion Torrent 318芯片在Ion Torrent PGM平台进行测序反应,共65个测序循环。
1.3.5 数据分析 按照参考文献[3-4],采用Ion Torrent Suite v3.0软件进行Ion Torrent数据提取、序列比对及SNPs和Indels提取,得到的SNPs和indels经dbSNP 137数据库过滤后,检索HGMD、LOVD等数据库及Pubmed相关文献,匹配已报道的致病位点,确定的致病突变及可疑致病突变均经Sanger法测序验证。检测MCCC1基因(NM_020166)19个外显子的编码区,共编码726个氨基酸,外显子编码区覆盖度:99.3%;检测MCCC2基因(NM_022132)17个外显子的编码区,共编码564个氨基酸,外显子编码区覆盖度:100%。基因诊断均由本实验室完成。
2 结果
2.1 新生儿筛查及基因检测结果 65 451例新生儿筛查中通过基因检测确诊3例MCCD患儿:MCCD患儿1例,父源性MCCD 1例,母源性MCCD 1例。另外还有1例患儿仅找到1个致病位点。
2.2 MCCD患儿和父源性MCCD检测结果 新生儿初筛时C4DC+C5OH为5.64 μmol/L,C0正常,召回复查后C4DC+C5OH为5.79 μmol/L,尿气相色谱质谱检测到高浓度的3-MCG和3-HIVA,符合MCCD临床诊断。患儿MCCC2基因检测到2个致病突变,均为c.659G>A纯合突变。在其父亲MCCC2基因上检测到2个致病突变,分别为c.659G>A突变和c.422A>G突变,其中c.422A>G突变导致氨基酸序列发生p.Lys141Arg突变。患儿母亲未检测到异常突变。综合基因检测结果,患儿及其父亲MCCD诊断明确,患儿的2个MCCC2等位基因c.659G>A突变,1个来源于父亲,1个为新发突变。见表1。
2.3 母源性MCCD患者检测结果 新生儿初筛时C4DC+C5OH为4.91 μmol/L伴C0降低,召回复查后C4DC+C5OH逐步下降,生后2月时下降76%,尿气相色谱质谱检测基本正常。该母源性MCCD患者血液中C4DC+C5OH为14.7 μmol/L伴C0降低,尿气相色谱质谱检测到高浓度的3-MCG和3-HIVA,符合MCCD临床诊断。在患儿母亲MCCC2基因上检测到2个致病突变,均为c.1025G>A纯合突变,新生儿MCCC2基因携带c.1025G>A突变,其父亲未检测到异常突变,综合基因检测结果,母源性MCCD诊断明确。见表2。

表1 MCCD患儿的串联质谱初筛、尿气相色谱质谱和基因检测结果
注:C4DC+C5OH参考范围为0.08~0.6 μmol/L;C0参考范围为10~50 μmol/L。

表2 母源性MCCD患者及子女的串联质谱、尿气相色谱质谱和基因检测结果
注:C4DC+C5OH参考范围为0.08~0.6 μmol/L;C0参考范围为10~50 μmol/L。
2.4 仅找到1个致病位点的MCCD疑似患儿检测结果 新生儿初筛时C4DC+C5OH为1.29 μmol/L伴C0降低,召回复查后C4DC+C5OH未下降,尿气相色谱质谱检测到高浓度的3-MCG和3-HIVA,符合MCCD临床诊断。只在患儿的MCCC1基因上检测到1个致病突变,为IVS6+2T>A突变。在患儿父亲MCCC1基因上检测到IVS6+2T>A突变,其母亲未检测到异常突变。该患儿定期至我院儿童保健中心体检,生长发育正常。定期复查MS/MS提示C4DC+C5OH除了2次低于0.6 μmol/L,其余4次复查均波动于0.9 μmol/L上下,临床仍然倾向MCCD可能。虽然该例患儿仅找到1个致病位点,但是由于MCCC1基因外显子编码区覆盖度为99.3%,不能排除另一突变隐藏于非编码区或其他不确定因素的可能。见表3。
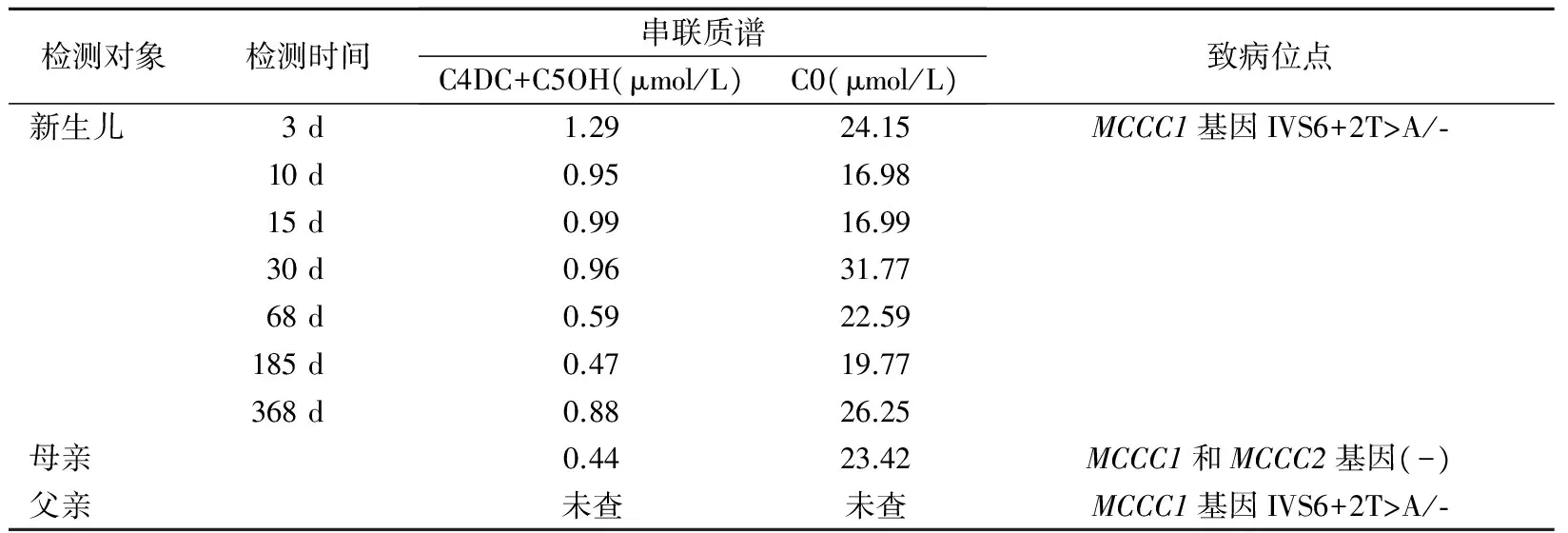
表3 MCCD疑似患儿的串联质谱、尿气相色谱质谱和基因检测结果
注:C4DC+C5OH参考范围为0.08~0.6 μmol/L;C0参考范围为10~50 μmol/L。
3 讨论
基因诊断是MCCD诊断的金标准,MCCD致病基因已明确为MCCC1和MCCC2,MCCC1定位3q25-27,含19个外显子,长度为2 580 bp,编码725个氨基酸;MCCC2定位5q12-q13.1,含有17个外显子,长度为2 304 bp,编码563个氨基酸,我院遗传医学中心针对MCCD设计的基因检测panel覆盖了MCCC1基因99.3%外显子编码区和MCCC2基因100%外显子编码区。
根据国内专家共识,MCCD的新生儿筛查和诊断已有相对规范化的程序,目前其争议和难点在于MCCD的临床分型。MCCD的临床表现差异很大,当前临床分型主要为:良性MCCD、恶性MCCD和母源性MCCD,其中,大部分为良性MCCD[5]。良性MCCD属于无症状型,临床上的处理措施主要为临床随访,定期复查血液中C4DC+C5OH和C0及尿液中3-MCG和3-HIVA浓度,如果随访中C4DC+C5OH异常升高并伴有C0降低时,补充左卡尼汀口服治疗。恶性MCCD病例目前国内未见报道,结合文献和相关书籍,认为恶性MCCD主要表现为呕吐、腹泻、抽搐、昏迷和肌张力异常等非特异性的消化道和神经系统症状,针对性的治疗措施尚存争议。第3种临床类型母源性MCCD,即母亲为MCCD患者,其血液中升高的C5OH可以通过母乳或胎盘传输给新生儿,导致其在新生儿筛查时血C5OH暂时性升高,脱离母体影响后逐步降至正常[6]。基于这部分新生儿的筛查结果,才能“回顾性”发现母亲是MCCD患者。
本研究中1例成年男性MCCD患者因为其后代的新生儿筛查而被间接检出并确诊,既往针对MCCD可疑阳性召回复查时多强调“母亲一同召回复查”,但是对于新生儿父亲的召回不够重视,通过本研究中母源性MCCD和新生儿父亲为MCCD病例的存在,提醒临床工作者遇到C5OH指标升高的新生儿一定要注意家系的MS/MS检测分析,而不能仅仅着眼于母亲复查。本研究中的MCCD患者拒绝定期复查MS/MS和GC/MS和补充左卡尼汀口服治疗,通过其家属随访至今暂无临床症状。该例父源性MCCD在父亲MCCC2基因上检测到2个致病突变,其中c.422A>G突变暂无相关报道,遗憾的是父亲的C4DC+C5OH检测结果暂缺,假设父亲C4DC+C5OH偏高,则倾向c.422A>G为可疑致病突变,相反,如果父亲C4DC+C5OH正常,那就可以间接判断c.422A>G是致病突变的可能就很小。
另外,本研究中还发现1例疑似MCCD患儿仅找到1个致病位点,这个病例中,患儿和父亲都具有IVS6+2T>A致病突变,患儿C4DC+C5OH持续偏高,遗憾的是患儿父亲的C4DC+C5OH检测结果暂缺,假设父亲C4DC+C5OH正常,那重点要考虑其他不确定的因素,例如本实验室采用的二代测序方法只能检测所检测基因范围内的点突变、微小缺失突变和微小插入突变,不能检测大片段缺失或插入突变,如果该患儿为大片段缺失,可进一步完善高密度寡核苷酸探针微阵列检测确诊。反之父亲C4DC+C5OH浓度与患儿接近,则倾向“杂合子”MCCD的可能。鉴于该患儿定期随访的MS/MS和GC/MS检测结果,临床仍倾向于MCCD,对于这一类人群,建议定期随访,如果随访中出现C0降低时,可以补充左卡尼汀口服治疗。
综上所述,新生儿筛查中C4DC+C5OH浓度升高的新生儿召回复查时建议一并对其家系,包括父亲和母亲均进行MS/MS检测分析,有利于发现和排除母源性[7]、父源性MCCD患者,同时临床上遇到一些非特异性的消化道和神经系统症状的患者应积极完善MS/MS检测,避免MCCD等遗传代谢病的漏诊。另外,必须指出,在临床工作中发现,MCCD患者由于暂无明显临床症状,患儿及家长的治疗依从性较差,容易失访,目前临床上倾向为良性MCCD的这部分患者后期是否会出现其他更为严重的临床表现无法预测,而且已有的研究均提示MCCD表型和基因型之间无明确的相关性,难以通过C5OH浓度[8]和基因型预测临床表型,所以这部分患者只能继续随访。不论是确诊的MCCD患者还是杂合子,当其伴有C0降低时,可以口服补充左卡尼汀治疗,因此,详细告知病情,定期随访和复查MS/MS、GC/MS很重要。
[1]Eldjarn L, Jellum E, Stokke O,etal. β-Hydroxyisovaleric aciduria and β-methylcrotonylglycinuia:a new inborn error of metabolism[J]. Lancet,1970, 2(7671):521-552.
[2]Frazier DM, Millington DS, McCandless SE,etal. The tandem mass spectrometry newborn screening experience in North Carolina:1997-2005[J]. J Inherit Metab Dis,2006,29(1):76-85.
[3]王彦云,孙云,杨冰,等. 应用新一代半导体靶向测序技术检测枫糖尿致病基因突变的研究[J]. 检验医学与临床, 2016,13(2):163-165.
[4]孙云,马定远,杨贵江,等. 一例瓜氨酸血症临床特征及ASS1基因突变分析[J]. 中华医学遗传学杂志, 2015,32(5):745-747.
[5]Baumgartner MR, Almashanu S, Suormala TC,etal. The molecular basis of human 3-methylcrotonyl-Co A carboxylase deficiency[J]. J Clin Invest, 2001,107(4):495-504.
[6]Cho KL, Kim YJ, Yang SH,etal. Maternal 3-methylcrotonyl-coenzyme A carboxylase deficiency with elevated 3-hydroxyisovalerylcarnitine in breast milk[J]. Korean J Pediatr, 2016,59(Suppl 1):S41-S44.
[7]Rips J, Almashanu S, Mandel H,etal. Primary and maternal 3-methylcrotonyl-CoA carboxylase deficiency: insights from the Israel newborn screening program[J]. J Inherit Metab Dis,2016,39(2):211-217.
[8]Forsyth R, Vockley CW, Edick MJ,etal. Outcomes of cases with 3-methylcrotonyl-CoA carboxylase (3-MCC) deficiency-Report from the Inborn Errors of Metabolism Information System[J]. Mol Genet Metab,2016,118(1):15-20.
(本文编辑:许晓蒙)
Clinical and genetic analysis of 4 child patients with 3-methylcrotonyl-coenzyme A carboxylase deficiency (MCCD) identified by neonatal screening
WANGYan-yun,SUNYun,CHENGWei,JIANGTao
(CenterforGeneticMedicine,MaternityandChildHealthCareHospitalAffiliatedtoNanjingMedicalUniversity,Nanjing210004,Jiangsu,China)
Objective To analyze 4 child patients with 3-methylcrotonyl-coenzyme A carboxylase deficiency (MCCD) identified by neonatal screening and confirmed by urine gas chromatography-mass spectrometry (GC/MS) and genetic analysis. Methods Newborns whose C4DC+C5OH concentration was above 0.6 μmol/L in newborn screening were recalled for rescreening, and the C4DC+C5OH concentrations in their mothers were detected. The child patients suspected with MCCD were further confirmed by urine GC/MS and genetic analysis. Results Three child patients were definitely diagnosed as MCCD by genetic analysis, including 1 MCCD, 1 maternal MCCD and 1 paternal MCCD. The other 1 child patient suspected with MCCD had only one allele in MCCC1. Conclusion The mother and father of newborns with elevated C4DC+C5OH identified in neonatal screening should routinely perform MS / MS testing. When only one pathogenic locus is found in the suspected MCCD child patients by genetic analysis, they should be followed up regularly.
3-methylcrotonyl-coenzyme A carboxylase deficiency; tandem mass spectrometry; 3-hydroxy-isovaleryl-carnitine; maternal; heterozygote
10.13602/j.cnki.jcls.2017.08.11
江苏省临床医学重点项目(BL2012039);江苏省卫生厅医学科研项目课题(H201343);南京市医学科技发展重点项目(ZKX14041);南京市科技发展计划(201405041)。
王彦云,1984年生,女,技师,硕士研究生,从事遗传代谢病检验专业。
蒋涛,主任技师,E-mail:jiangzhang784@163.com。
R446.1
A
2017-04-27)
猜你喜欢
肝博士(2022年3期)2022-06-30
食品安全导刊(2021年20期)2021-08-30
中老年保健(2021年9期)2021-08-24
临床肝胆病杂志(2020年5期)2020-12-20
城乡建设(2019年16期)2019-08-28
畜牧兽医科学(2018年12期)2018-02-18
畜牧兽医科学(2018年20期)2018-02-15
畜牧兽医科技信息(2018年5期)2018-02-13
特产研究(2014年4期)2014-04-10
中国环境科学(2014年4期)2014-02-02
