
熵在非晶材料合成中的作用∗
2017-09-09 01:02李蕊轩张勇
物理学报 2017年17期
李蕊轩 张勇
(北京科技大学,新金属材料国家重点实验室,北京 100083)
熵在非晶材料合成中的作用∗
李蕊轩 张勇†
(北京科技大学,新金属材料国家重点实验室,北京 100083)
(2017年6月1日收到;2017年6月21日收到修改稿)
熵作为系统的状态函数,对于真实物质体系而言是一个极为重要的物理量.在非晶态合金的制备过程中最具代表性的指导原则有“混乱原理”和井上三原则,二者皆与熵有着紧密的联系.在过去很长一段时间内,这些经验准则指导了大量新型非晶体系的发现,但近些年的实验结果对这些理论提出了质疑.除组元数目之外,还有其他尚待研究的因素也影响着合金体系的玻璃形成能力.本文总结了玻璃转变过程中熵在热力学条件、动力学条件和结构条件中所扮演的角色,阐述了其对玻璃形成能力产生的或正或反的影响.特别是对近几年发展起来的高熵非晶体系的研究有助于开发出临界尺寸更大的非晶合金,也有助于进一步探索多组元合金和非晶形成能力之间的关系.
熵,非晶形成能力,高熵非晶,块体非晶
1 引 言
20世纪30年代初,通过多次蒸发沉积实验,德国物理学家Kramer[1]制备出了Sn的非晶态金属薄膜,这是人类首次发现非晶态金属.1952年,Turnbull通过水银过冷实验总结得出提高冷却速度可以使得金属熔体达到远离平衡态的深过冷温度区间而不发生结晶,冷却速度越快这一现象越明显;随后他又提出,对于任何成分的金属熔体,只要冷却速率足够快,都可以以一种无扩散的方式凝固而形成玻璃[2].因此,在这一领域,Turnbull是公认的非晶合金的理论奠基人.1960年,这一猜想得到证实,加州理工学院的Duwez及其团队[3]利用冷却速率高达106K/s的快冷技术制备出了厚度仅有 20µm的Au75Si25非晶合金样品,这是史上第一次用人工的方法得到的真正意义上的非晶合金.然而,传统合金体系的玻璃形成能力通常较弱,必须通过极高的冷却速率来满足制备需求,这在很大程度上阻碍了金属玻璃的进一步发展.
人们并未停下探索未知世界的脚步,研究人员一直以来尝试各种办法制备更大尺寸的非晶态合金.20世纪60年代末,陈鹤寿[4]研究了钯基合金体系的玻璃形成能力,第一次制备出直径为毫米级的Pd-Cu-Si金属玻璃棒.1989年,日本东北大学的井上明久等[5,6]采用非贵金属元素制备出毫米级金属玻璃Mg-Cu-Si和La-Al-Ni.随后性能优良的Zr,Zn,Cu,Sr,Ti等非晶体系也相继问世[7−9],其临界尺寸均超过了1 mm,对制备条件的要求不再苛刻.此后,三维尺度在毫米量级的金属玻璃被称为块体金属玻璃(bulk metallic glass,BMG).20世纪90年代初,Greer提出“混乱原理”,在这一原理的启迪下,日本和美国的两个课题组几乎同时制备出多组元的块体金属玻璃,临界尺寸达到厘米级,并将临界冷却速率减小至1 K/s[10−14].井上明久在实践的基础上不断归纳总结出块体非晶合金的形成规律,提出了高玻璃形成能力的合金体系应该遵守三个准则:1)合金体系由至少三个元素组成;2)3个主要的组成元素必须具有足够大的原子尺寸差;3)组成的组元之间应当存在较高的混合放热值.在这以后,块体非晶的发展迎来高峰期,相继制备出了不同成分的块体金属玻璃.
然而,2004年Cantor等[15]基于实验结果对Greer的混乱原理提出质疑,他最多合成了含有20种金属元素的合金体系,但对这些合金进行铸造和熔融纺丝都不
能获得非晶态结构,因此他指出混乱原理并不能完全描述非晶态结构形成的规律,其他因素也发挥着重要作用.
在统计物理中,熵(S)是一个用来描述体系混乱程度的物理量,其表达式为

其中Ω为在宏观态中包含微观态的总数,k为玻尔兹曼常数.在热力学中,克劳修斯则给出在可逆过程中熵的变化:

其中δQ是在此过程中体系与外界所交换的热量.
一般而言,一个真实的物质体系的熵包括构型熵Scon,振动熵Svib,电子熵Selec和磁性熵Smag,因此,

在金属和合金体系的总熵中,电子熵和磁性熵所占比例一般很小.因此,(3)式可简化为

一般而言,金属玻璃样品在恒压条件下制备,因此用吉布斯自由能(G=H−TS)来描述、判断整个过程是非常方便的.随着吉布斯自由能不断减小,描述微观状态的熵S和描述体系内能的焓H将协同作用,在不同的温度范围内,二者应该有着分明的主次关系.本文突出强调熵在整个玻璃转变过程中所起的作用,同时阐述了其在近些年新发展的高熵金属玻璃中的地位.
2 非晶形成的热力学条件
过冷液体的晶化包括两个方面:一是晶核的形成,涉及固-液两相的自由能差和二者界面能的相对关系,与构型熵和振动熵密切相关;二是晶核的长大,涉及原子的扩散能力.当温度低于熔点Tm时,合金熔体进入过冷液相区,随着温度进一步降低,过冷度逐渐增大,此时,从过冷液体到晶态固体的相变驱动力亦逐渐增大,形核临界半径逐渐减小,这意味着形核愈来愈容易,但是原子的扩散随着温度的降低而变得困难,晶核的长大被抑制.因此,通常在某一温度区间形核与生长能够相互妥协而大大缩短晶化时间,即在这一最优温度区间,既能形核又能使晶核快速长大.从形成金属玻璃的角度来看,一方面要尽量缩短熔体在这一温度区间所停留的时间,另一方面要延长晶化时间.前者与冷却速率有关,主要取决于实验手段、样品尺寸和合金本身的导热能力,后者则与合金相变驱动力、固液两相界面能和原子的扩散系数相关.从凝固形核角度考虑,在过冷液体中能够稳定存在的临界形核半径rc为

其中ΔGl-s为液固两相的自由能差,σ为两相界面能.那么,形成此晶核所需的临界能量Ec为

显而易见,临界半径rc越大,临界能量Ec越大,晶化越不容易发生,也即越容易形成玻璃态.要想获得较大的临界能量,增加玻璃形成能力,就要减小固液两相之间的自由能差.
根据Thompson提出的凝固过程中固液两相自由能差的公式:

其中ΔT表示过冷度,ΔSf表示熔化熵,也就是固液两相在熔点附近的熵值差.由(7)式可知过冷度越大,固液两相吉布斯自由能差ΔGl-s越大,此时容易形成晶体.但如果减小固液两相之间的熵值差ΔSf,就会使ΔGl-s减小,在这种情况下就更容易形成非晶体.
相对于晶态固体,合金的过冷熔体因为其能量更高,原子分布更加混乱,所以液态的熵值通常较大.在液体状态中,因为原子处于较高能量状态,可以认为由温度主导的振动熵对总熵的贡献较大,而较大的原子间距使得构型熵的作用很小,所以液态时多组元合金和单质金属的熵值相差无几.当温度降低,熔体凝固后,原子基本被限制在平衡位置附近,振动熵大大减小,而构型熵在总熵中所占比例增大,因此多原子排列组合的构型使得构型熵的作用在竞争中更明显,即多组元合金的熵比单组元金属的熵高得多.就此意义而言,在多组元合金中液固两相熵值差减小,凝固过程中的某一瞬时中,熔体中的原子排布状态就与液态中的原子排布状态更为接近,此时过冷液体和结晶相的吉布斯自由能差ΔGl-s较小,所以就有利于非晶的形成,即多组元高熵合金体系的非晶形成能力更强,如图1所示.

图1 (a)总熵值较小;(b)总熵值较大的合金体系液固两相原子的排布状态Fig.1.The atomic arrangement of liquid and solid phases in(a)low-entropy alloy,(b)high-entropy alloy.
由剑桥大学的Greer提出的“混乱原理”在很长一段时间内指导了大量高玻璃形成能力的合金体系的发现,即合金组元越多,玻璃形成能力越强.这就是基于热力学条件下熵对非晶形成过程的促进作用.在熵的玻尔兹曼定义式中,N种元素组成的理想熔体的构型熵可以表达为

其中ci表示原子分数,R表示理想气体常数.假设当合金为等原子比时,各元素的原子分数都相等,也就是说ci=1/N,因此

即组元越多,混合熵越高.尽管“混乱原理”没有直接说明各原子的相对含量,但表示了多组元和高混合熵对非晶态形成的促进作用.
非晶态是一种亚稳态,无时无刻不在趋向稳定态,如最初合成的Au75Si25非晶态薄膜只能在室温下存在几个小时.因此,形成非晶除了要提高熔体冷却过程中的玻璃形成能力之外,还要提高非晶相的稳定性.研究表明,非晶的稳定性与其玻璃形成能力密切相关[10],具有高稳定性的非晶态体系通常具有高的玻璃形成能力和宽的过冷液相区.
所以从热力学上讲,可以通过熵值的增加来提高玻璃形成能力和非晶的稳定性.也就是说增加熵值有利于非晶的形成.
3 非晶形成动力学条件
从热力学上讲,大过冷度下相变驱动力极大,过液体变得极不稳定.但是热力学只是非晶形成的必要条件,动力学对非晶的形成和抑制晶化也至关重要.单组元金属的晶化是一个同素异型转变,没有成分的变化,再加上金属键无方向性,原子只需要在近程调整位置即可晶化,所以此类晶化过程几乎无法抑制.而对于二元甚至多元合金,结晶相和液相成分差别大,又有多相竞争生成,晶化的形核和生长需要原子做长程运动,因此晶化过程容易从动力学上被抑制,有利于非晶态合金的生成.
形成非晶是一个避免非均匀形核和推迟均匀形核的过程,这一过程可以通过控制两个方面来实现,即提高冷却速度和改变合金体系.
根据非晶态合金形成的动力学原理,对于给定的合金熔体,当温度降至熔点Tm以下时,它可以通过两种途径凝固(如图2所示):当冷却速度R<Rc(如R1)发生晶化转变,需要形核与长大过程;而当R>Rc(如R2)时,形核和长大过程没有充分的时间,熔体进入过冷状态,随着温度的进一步降低,液体黏度将会逐渐升高到1012Pa.s,或者弛豫时间达到102s.此时,粒子在有限的时间内很难改变位置形成新的构型,从而导致体系偏离平衡态,最终进入了非晶态.

图2 过冷液体连续冷却转变曲线Fig.2.TTT curves of the supercooled liquid.
最初使用提高冷却速度的方法获得非晶,当冷却速度高于鼻尖温度所对应的冷却速度,就能抑制非均匀形核并阻止晶核长大.加州理工学院的Duwez及其同事获得的Au75Si25非晶合金样品就是利用冷却速度达到106K/s的快冷技术制备成的.后来发展的甩带法等制备非晶条带的技术也是依照这一原理.
现在,通过调整合金体系成分可以在较低冷却速度下获得大块非晶.从晶核生长方面看,如果晶核长大过程中熔体的黏度较大,原子扩散能力就差,在凝固时原子就不容易长程扩散从而进行形核、长大,这时就容易克服热力学熵的阻力而获得非晶形成的动力学优势,得到原子排列混乱的、短程有序的非晶体.
熵和黏度的关系使其对于非晶的形成和稳定非常重要,液体的构型熵和黏滞系数η的关系可用Adam-Gibbs模型描述:
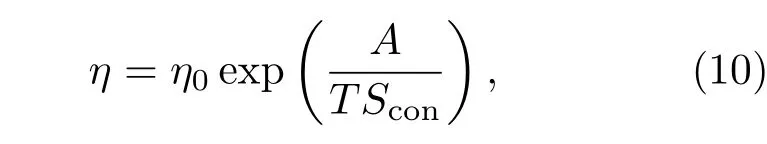
其中A为常数,η0为一个大气压下的参考动力学黏滞系数.可见,构型熵越高,熔体的黏滞系数越低.熔体的流动在微观上可以理解为原子排布的一个构型向另一个构型的转变,这一转变越容易、进行的速度越快,熔体的流动性越好、黏度就越小.当熔体的构型熵增加,也就是熔体中原子可能存在的构型数目增加时,不同构型之间的转变就会变得容易,因而使得熔体流动性增强,黏度减小.再根据Stokes-Einstein关系描述的液体的黏度和有效扩散系数的关系

其中a为原子间距,η表示液体的黏滞系数.熔体黏滞系数的降低导致扩散系数增大,原子充分扩散和重新排列的机会增大,原子长程扩散使得晶核形成和长大的可能性增大,此时倾向于形成有序的金属间化合物,而不是非晶体.
因此,从非晶形成的动力学条件上讲,高熵合金体系的玻璃形成能力较差.
4 非晶形成的结构条件
在构型熵中还包含错配熵Sσ,通常使用标准化错配熵Sσ/kB来描述体系中原子的错配度,反映体系中弹性能的大小.大量玻璃形成体系的计算表明,随着Sσ/kB的增加,混合焓ΔHmix减小,较大的错排度使得原子倾向于混乱随机排列,原子间应力就较大,从而使得有序相不稳定,易于表现出无序排列的非晶结构.例如将原子尺寸很小的铍元素加入锆基合金中将大幅提高玻璃形成能力.错配熵的大小可能还与晶化相复杂程度相关.单质硅等非金属元素结晶态下共价键的旋转,使得它比由金属键结合的单质金属复杂程度要高,所以单一组元的Si,Sn等比单质金属更容易形成非晶,以此类比至多组元合金中,随着Sσ的增加,我们可以认为晶化相由于原子相差较大而变得更加复杂,此时即使在过冷液体中原子扩散速率很快,也不容易发生结晶.因此,可以使用错配熵值来预测玻璃形成能力,高的玻璃形成能力的合金体系Sσ/kB临界值是0.1,而非晶体系中的平均值可以达到0.33[16].
已有计算表明Sσ/kB~(δ/22)2[17],可见错排熵与原子尺寸差δ有关,原子尺寸差越大,错排熵越大.从非晶形成的结构条件来看,错排熵的提高有利于非晶形成能力的提高,这也与井上经验准则中要求的“组成元素之间必须具有足够大(>12%)的原子尺寸差”相一致.
除错排熵外,还可以将与原子尺寸和原子堆垛比率有关的结构因素导致的熵值的变化统一归结成SE,这相当于实际硬球混合模型与理想固溶体的差,此时总熵等于构型熵加由结构因素导致的额外构型熵,即S=Scon+SE.计算表明,在能够形成非晶相的合金体系中,随着原子堆垛比率的增加,SE的值明显增大,而在固溶体体系中SE的值相对较小[18].
因此,不管是用Sσ或是SE衡量结构因素,均表现为熵值越大,非晶形成能力越强.
5 高熵非晶
根据最初的非晶形成“混乱原理”和井上三原则,由多组元的等原子比或近等原子比金属元素组成的合金体系可能具有优良的非晶形成能力.台湾清华大学Yeh等[19]于2004年首次提出高熵合金的概念.高熵合金是具有5种及5种以上等原子比成分的合金体系,它表现出四大突出效应:高混合熵效应,迟滞扩散效应,晶格畸变效应和鸡尾酒效应.人们一开始认为,从已有的非晶形成理论方面讲,高熵合金天然满足多组元的要求,且近等原子比的成分使得溶质溶剂的区分并没有那么明显,复杂的成分使得凝固瞬间满足晶体相的成分的形成要求比较困难,严重的晶格畸变和迟滞扩散效应阻碍了原子扩散,所以高熵合金体系理论上存在形成非晶相的倾向.但是实验证明,高熵合金通常会形成以简单的面心立方或体心立方为主的固溶体结构.因为高熵效应在玻璃形成过程中多面性的影响,高熵合金的玻璃形成能力并没有人们期待的那么大.

图3 高熵非晶的概念图Fig.3.A concept map of high-entropy-glass.
如图3所示,高熵非晶是高熵合金和非晶态合金集合的交集. 2011年,汪卫华课题组[20]第一个提出了这一概念并制备出了块体Sr20Ca20Yb20(Li0.55Mg0.45)20Zn20高熵非晶,它具有极低的玻璃转变温度并能在室温下表现出类似于高分子的热塑性变形行为. 接着,Takeuchi等[21]首次制备出了同时含有金属和非金属元素的高熵非晶Pd20Pt20Cu20Ni20P20.此后,人们又开发出多种具有良好性能的高熵非晶体系. 如具有良好生物相容性的Ca20Mg20Zn20Sr20Yb20[22],提高了移植生物体内的降解性;Fe25Co25Ni25(PxC0.8−xB0.2)25具有良好的软磁性能[23],矫顽力仅为1.2 A/m;GeNbTa-TiZr具有良好的热稳定性,在750°C经1 h退火后仍能保持非晶态结构[24].表1总结了到目前为止发现的所有高熵非晶.一开始人们希望通过替换或增加周期表中的同族或化学性质相近的元素,由已有的非晶形成能力强的体系获得高熵非晶体系,如高熵非晶Pd20Pt20Cu20Ni20P20和Ti20Zr20Cu20Ni20Be20分别是由已有的非晶合金
体系Pd40Ni40P20和Zr41.2Ti13.9Cu12.5Ni10Be22.5通过相似元素替换满足等原子比要求而实现的.替换后合金体系熵值增加,但这并不表示玻璃形成能力也会增加,相反,实验证明玻璃形成能力在替换元素后大大减小.如Pd40Ni40P20体系临界直径为25 mm,而高熵非晶Pd20Pt20Cu20Ni20P20[21]的临界直径只有10 mm.Ti20Zr20Cu20Ni20Be20[25]体系非晶的临界直径仅为3 mm,虽然经过研究添加Hf后可以提高玻璃形成能力,Ti20Zr20Hf20Be20(Cu7.5Ni12.5)的临界直径也只能增加至30 mm[26].可见已有的高熵非晶体系玻璃形成能力都不高,所以最佳的非晶形成体系并不一定适用于高熵范围.

表1 目前为止发现的高熵非晶Table 1.High-entropy-glass systems discovered till now.
2008年,北京科技大学张勇等[33]总结了多组元合金体系的相形成规律.多组元合金形成固溶体相或者非晶相由固有原子尺寸差δ和化学混合焓ΔHmix共同决定.定义原子尺寸分散度和混合焓分别为


其中Ωij=4Δ是二元液态等原子合金的混合焓.当δ足够大或者ΔHmix是足够大的负数时,就倾向于形成非晶相[34].在较负的ΔHmix下,除了可能形成非晶相外,还可能形成长程有序的金属间化合物,此时需要考虑影响原子排布的构型熵Scon,也有文献将构型熵称作混合熵,用ΔSmix表示. 绘制ΔHmix-δ图和ΔSmix-δ图,可以发现非晶相的形成区域为δ≥9,−49≤ΔHmix≤−5.5(kJ/mol),7≤ ΔSmix≤ 16(J/(K.mol)),也就是说,当所涉及的合金成分参数落在这一范围内时,多组元合金才会形成非晶.Li等[23]通过计算证实12种已有的高熵非晶体系的参数均落在这一范围内,这间接证明了这一预测的准确性.
6 结 论
非晶态结构的形成是熔体冷却过程中多相竞争、形核长大过程被充分抑制的结果,这一过程的影响因素多且复杂.从熵的角度讲,它对非晶形成的作用具有多面性,如图4所示.从非晶形成的热力学条件和结构条件而言,熵值的增加通过分别作用于固液两相自由能差和原子尺寸差而使玻璃形成能力提高,但从动力学条件而言,熵值增加会使黏度减小,因而使玻璃形成能力降低.所以,单纯基于多组元高混合熵的“混乱原理”并不能完全描述玻璃形成能力,多组元高熵合金在不同条件下可以形成固溶体、金属间化合物或非晶相,高熵非晶只是其中的一部分,只有满足由原子尺寸差、混合焓和混合熵限定的成分区域才能形成非晶相.对高熵非晶体系的研究有助于进一步探索多组元合金和非晶形成能力之间的关系,从而进一步提高非晶形成能力.

图4 熵在非晶形成过程中的多方面作用Fig.4.A schematic about the in fl uence on glass forming ability by entropy.
感谢日本东京大学王建国博士的有益建议和讨论.
[1]Kramer J 1934 Annln.Phys.19 37
[2]Johnson W L 1986 Prog.Mater.Sci.30 81
[3]Klement W,Willens R,Duwez P 1960 Nature 187 869
[4]Chen H S 1974 Acta Metall.22 1505
[5]Inoue A,Kato A,Zhang T,KiMS G,Masumoto T 1991 Mater.Trans.JIM32 609
[6]Inoue A,Kita K,Zhang T,Masumoto T 1989 Mater.Trans.JIM30 722
[7]Inoue A,Zhang T,Masumoto T 1990 Mater.Trans.JIM31 177
[8]Jiao W,Zhao K,Xi X K,Zhao D Q,Pan MX,Wang W H 2010 J.Non-Cryst.Solids 356 1867
[9]Li H F,Zhao K,Wang Y B,Zheng Y F,Wang W H 2012 J.Biomed.Mater.Res.B:Appl.Biomater.100 368
[10]Wang W H,Dong C,Shek C H 2004 Mater.Sci.Eng.R 44 45
[11]Peker A,Johnson W L 1993 Appl.Phys.Lett.63 2342
[12]Inoue A,Nakamura T,Nishiyama N,Masumoto T 1992 Mater.Trans.JIM33 937
[13]Inoue A,Zhang T,Masumoto T 1989 Mater.Trans.JIM30 965
[14]Inoue A,Zhang T,Nishiyama N,Ohba K,Masumoto T 1993 Mater.Trans.JIM34 1234
[15]Cantor B,Chang I T H,Knight P,Vincent A J B 2004 Mater.Sci.Eng.A 375–377 213
[16]Takeuchi A,Inoue A 2000 Mater.Trans.JIM41 1372
[17]Takeuchi A,Amiya K,Wada T,Yubuta K,Zhang W,Makino A 2013 Entropy 15 3810
[18]Samaei A T,Mohammadi E 2015 Mater.Res.Express 2 096501
[19]Yeh J W,Chen S K,Lin S J,Gan J Y,Chin T S,Shun T T,Tsau C H,Chang S Y 2004 Adv.Eng.Mater.6 299
[20]Zhao K,Xia X X,Bai H Y,Zhao D Q,Wang W H 2011 Appl.Phys.Lett.98 141913
[21]Takeuchi A,Chen N,Wada T,Yokoyama Y,Kato H,Inoue A,Yeh J W 2011 Intermetallics 19 1546
[22]Li H F,Xie X H,Zhao K,Wang Y B,Zheng Y F,Wang W H,Qin L 2013 Acta Biomater.9 8561
[23]Li Y,Zhang W,Qi T 2017 J.Alloy.Compd.693 25
[24]Cheng C Y,Yeh J W 2016 Mater.Lett.181 223
[25]Ding H Y,Yao K F 2013 J.Non-Cryst Solids 364 9
[26]Ding H Y,Shao Y,Gong P,Li J F,Yao K F 2014 Mater.Lett.125 151
[27]Gao X Q,Zhao K,Ke H B,Ding D W,Wang W H,Bai H Y 2011 J.Non-Cryst.Solids 357 3557
[28]Huo J,Huo L,Men H,Wang X,Inoue A,Wang J,Chang C,Li R W 2015 Intermetallics 58 31
[29]Zhao S F,Yang G N,Ding H Y,Yao K F 2015 Intermetallics 61 47
[30]Qi T,Li Y,Takeuchi A,Xie G,Miao H,Zhang W 2015 Intermetallics 66 8
[31]Zhao S F,Shao Y,Liu X,Chen N,Ding H Y,Yao K F 2015 Mater.Design 87 625
[32]Cheng C Y,Yeh J W 2016 Mater.Lett.185 456
[33]Zhang Y,Zhou Y J,Lin J P,Chen G L,Liaw P K 2008 Adv.Eng.Mater.10 534
[34]Guo S,Hu Q,Ng C,Liu C T 2013 Intermetallics 41 96
PACS:71.23.Cq,71.55.Jv,78.55.QrDOI:10.7498/aps.66.177101
*Project supported by the National Natural Science Foundation of China(Grant Nos.51471025,51671020).
†Corresponding author.E-mail:drzhangy@ustb.edu.cn
Entropy and glass formation∗
Li Rui-Xuan Zhang Yong†
(State Key Laboratory for Advanced Metals and Materials,University of Science and Technology Beijing,Beijing 100083,China)
1 June 2017;revised manuscript
21 June 2017)
Entropy is a state function of the real physical system,which relates to the chaos of a system.During the longterMexploring glass-forming systems,many empirical rules are put forward,including “confusion principle” and three empirical rules.
Over a long period of exploring,many glass-forming alloys are developed based on those principles,while some questions have been raised in recent years based on the experimental results,because some other uncertain factors also have in fl uence on the glass-forming ability(GFA)except a number of constituents,e.g.,entropy.Greer claimed that in the “confusion principle” the higher the entropy value,the better the glass-formation ability will be,which does not accord with the recent experimental results.
The e ff ects of entropy on the glass-formation ability are summarized froMthe viewpoints of thermodynamics,kinetics,and complexity of atomic structures.In the aspects of thermodynamics and structure,the increase of entropy has a positive e ff ect on glass formation,while in kinetics,the in fl uence is negative.FroMthe viewpoint of thermodynamics,the increase of entropy leads to the decrease of the entropy di ff erence between solid phase and liquid phase,and therefore,the di ff erence in Gibbs free energy between these two phases decreases.At a certain time during solidi fi cation,compared with the low-entropy alloy,the high-entropy alloy in the solid phase has an atomic arrangement close to that in the liquid,and it is more likely to forMthe amorphous phase.
In the aspect of kinetics,the increase of entropy results in the decrease of the viscosity of the systeMaccording to the Adam-Gibbs equation.As a result,atoms di ff use easily in the systeMand the ordered-phase is more likely to form,which means that the glass-formation ability decreases with the increase of entropy.Furthermore,in the aspect of atomic structure,the increase of mismatch entropy relates to the big mis fi t degree between atoms,i.e.,the large atomic size di ff erence.Atoms in the high-entropy alloy tend to distribute disorderly in the system,and therefore the stress between atoms increases.As a result,with the increase of the entropy,the ordered-phase becomes unstable and the GFA will be enhanced.
Furthermore,the high-entropy-glass is brie fl y reviewed and analyzed,which is a new systeMbetween high-entropy alloy and amorphous alloy.There have been many high-performance high-entropy-glass systems reported up to now.Researches about this unique systeMwill contribute to developing some new amorphous alloys with excellent performances,and more importantly,to exploring the complex relationship between GFA and multicomponent alloys.
entropy,glass-forming ability,high-entropy-glass,bulk metallic glass
10.7498/aps.66.177101
∗国家自然科学基金(批准号:51471025,51671020)资助的课题.
†通信作者.E-mail:drzhangy@ustb.edu.cn
©2017中国物理学会Chinese Physical Society
http://wulixb.iphy.ac.cn
猜你喜欢
上海金属(2022年4期)2022-08-03
民族文汇(2022年14期)2022-05-10
贵州大学学报(自然科学版)(2021年5期)2021-09-26
机械制造(2020年5期)2020-02-20
高教学刊(2019年9期)2019-09-10
精密成形工程(2018年6期)2018-11-23
湖南大学学报·自然科学版(2017年12期)2018-01-17
中国科技纵横(2016年19期)2016-12-10
山东工业技术(2016年15期)2016-12-01
中国科技信息(2015年21期)2015-11-07
