
奥美沙坦酯关键中间体中的杂质结构分析
2017-09-03 10:56李红
分析仪器 2017年4期
李 红
(辽宁省分析科学研究院 辽宁省标准化体系建设工程技术研究中心,沈阳 110015)
奥美沙坦酯关键中间体中的杂质结构分析
李 红
(辽宁省分析科学研究院 辽宁省标准化体系建设工程技术研究中心,沈阳 110015)
研究了奥美沙坦酯关键中间体8中的主要杂质结构,先通过液质联用分析初步推测其可能结构,然后以二氨基马来腈和原丁酸三甲酯为原料,经5步反应合成了其可能杂质7a和7b,采用核磁共振氢谱和NOE确定了7a和7b这一对同分异构体各自的分子结构,并通过HPLC保留时间比对确认了奥美沙坦酯关键中间体中的主要杂质为7a,该研究结果为奥美沙坦酯质量控制提供了很好的借鉴。
奥美沙坦酯关键中间体 杂质 结构分析
奥美沙坦酯是由日本三公株式会社开发成功的新的AngⅡ受体拮抗剂。奥美沙坦酯是前药,其代谢产物奥美沙坦才是生理活性的药物。奥美沙坦酯与其他沙坦类药物相比,具有对AT1受体的选择性作用高,能使舒张压和收缩压在24h内持续平稳降低,故显示出强效和长效的作用,且副作用少,是目前上市的沙坦类药物中总体疗效较好的品种[1-5]。在奥美沙坦酯原料药中,现有研究已发现其中有15种杂质,这些杂质的结构均经过全合成或谱学等手段确认。然而,这些杂质来源于何种中间体以及引入方式尚未有细致的研究,因此,研究其关键中间体中的杂质结构和含量对最终药物的质量控制具有重要的意义。
化合物8(结构如图1所示)是制备奥美沙坦酯的关键中间体,目前市场上该工业品的含量在96%左右,尚含有4%的杂质。而关于这4%杂质的类型以及是否会引入最终产物中尚未有细致的研究。本实验在对市场上销售的化合物8的含量分析过程中,发现其中有一个杂质的含量约为2%,其它杂质的含量都在0.5%以下,而该杂质的结构尚未有文献报道。实验在参考化合物8的合成工艺的基础上并结合超高液相色谱和质谱联用分析,推测该杂质可能为7a或7b(如图2所示)。
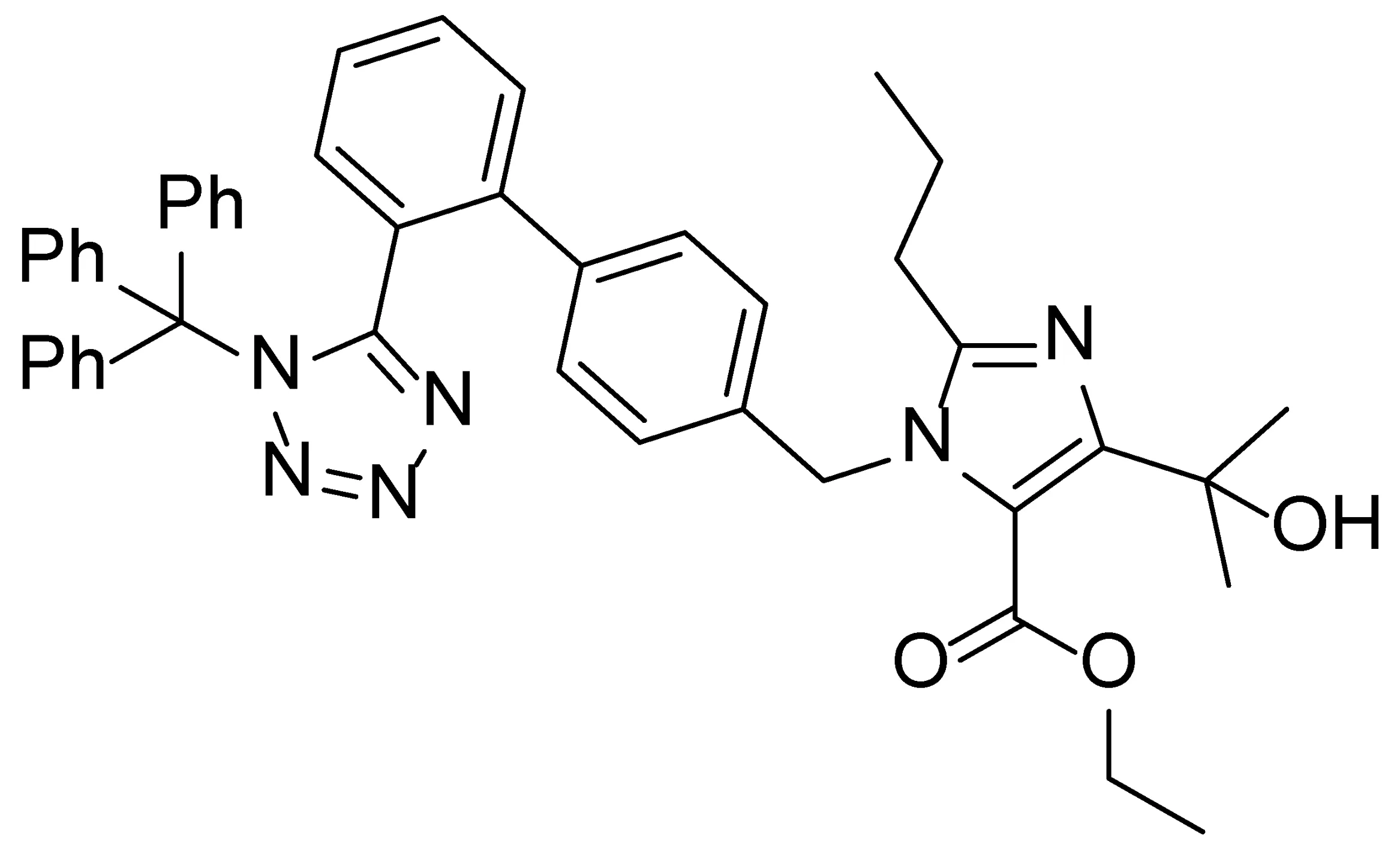
图1 奥美沙坦酯关键中间体8
因此,实验在参考文献方法的基础上以二氨基马来腈和原丁酸三甲酯为原料,经过缩合环化得化合物2、化合物2与格式试剂反应得化合物3、化合物3的氰基经水解后酯化得到化合物6,化合物6与联苯溴苄衍生物反应得化合物7a和7b, 并采用谱学手段确定了其各自的结构,并与化合物8中的杂质进行液相色谱分析比对,发现其中的杂质为化合物7a。7a在后续的反应中生成了原料药中的杂质C。因此,控制中间体8中杂质7a的含量将对控制奥美沙坦酯产品质量具有重要价值[6-10]。具体合成路线如图2所示。
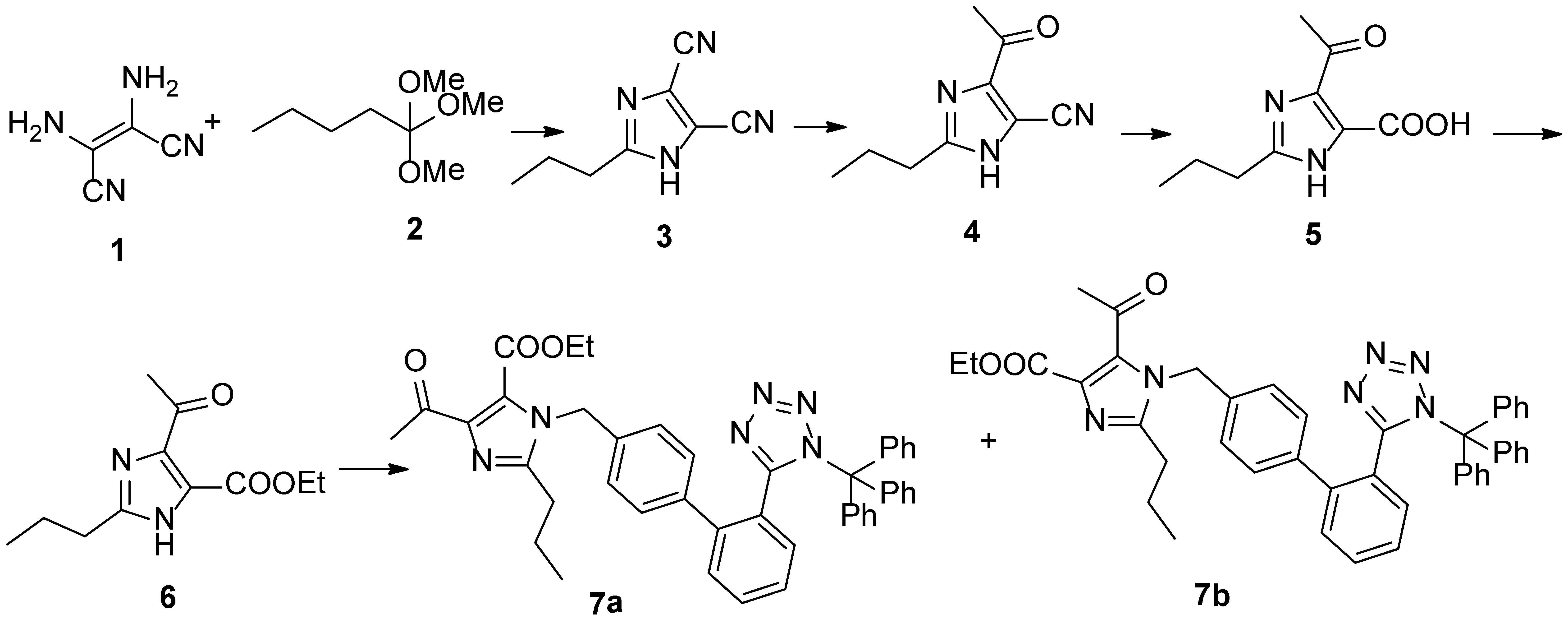
图2 目标产物化合物7a的合成
1 实验部分
1.1 试剂与仪器
Bruker NMR 500MHz(CDCl3或DMSO为溶剂,TMS为内标,德国布鲁克公司);Ultima Global Spectrometer型质谱仪(ESI源,美国Waters公司);RY-1显微熔点仪。
薄层层析用硅胶板、柱层析用硅胶(200~300目,青岛海洋化工厂);如果未经注明,使用的试剂均为分析纯,未做进一步纯化。
1.2 合成方法
1.2.1 化合物3的合成
将二氨基马来腈(24.1 g, 0.162 mol)溶于乙腈(300 mL)中,加入原丁酸三甲酯(26.4 g, 0.178 mol)。所得反应混合物在回流温度下搅拌5小时, TLC检测原料反应完全后减压浓缩回收乙腈。将残余物用二甲苯(87 mL)稀释,所得溶液在回流温度下搅拌7小时。冰浴中冷却后,析出大量棕色固体、过滤、滤饼经少量二甲苯洗涤后干燥得25.4 g化合物3的纯品,产率为98.0%。熔点:141~144℃。熔点数据与文献[4]报道一致。
1.2.2 化合物4的合成
在氮气氛围下,将化合物3 (20 g, 0.13 mol)溶于干的THF (200 mL)中,冰浴冷却下滴加1M MeMgCl (160 mL, 0.16 mol)的四氢呋喃溶液,控制滴加速度使反应液温度不超过20℃。滴加完毕后,所得反应混合物在室温下继续搅拌反应0.5小时,TLC检测原料反应完全后,加入饱和氯化铵溶液(200 mL)继续搅拌2小时以终止反应,然后加入乙酸乙酯(200 mL),搅拌均匀后加入硫酸氢钾,分出有机相,水相用100 mL乙酸乙酯萃取一次。合并有机相,有机相经饱和食盐水洗涤后无水硫酸钠干燥,过滤,滤液减压浓缩至干,得固体化合物4粗品,所得粗品在石油醚/乙酸乙酯中重结晶得19.5 g化合物4的纯品。产率为88.0%。熔点:93~94℃。熔点与文献[4]报道一致。1H-NMR(CDCl3, 500 MHz),δ: 1.00 (t,J=7.5 Hz, 3H); 1.81 (m,J= 7.5 Hz, 2H); 2.72 (s, 3H); 2.79 (t,J=7.5 Hz, 2H); 10.70 (brs, 1H)。MS(ESI+),m/z:178.2[M+H]+。所得谱学数据与文献[4]报道一致。
1.2.3 化合物6的合成
将化合物4(4.00 g,23.2mmol)溶于6N HCl(60mL)溶液中。所得反应混合物在回流温度下搅拌反应8小时, TLC检测原料反应完全后减压浓缩至干,加入100 mL乙醇后浓缩至干,重复3次得化合物5的粗品。所得化合物5粗品无需纯化,直接用于下一步的酯化。
将化合物5溶于EtOH(100mL)中。在室温下向该乙醇溶液中通入HCl气体20分钟。所得反应液在室温搅拌反应16小时后,将溶液真空浓缩。所得残余物溶于乙酸乙酯和碳酸氢钠的混合溶液中。分出有机相,水相用100 mL乙酸乙酯萃取1次。合并有机相,有机相经饱和食盐水洗涤后无水硫酸钠干燥,过滤,滤液减压浓缩至干,得固体化合物6粗品,所得粗品在石油醚/乙酸乙酯中重结晶得3.5 g化合物6的纯品。产率为70.0%。熔点:80.5-82℃。1H-NMR(CDCl3, 500 MHz),δ: 0.98 (t,J=7.5 Hz, 3H); 1.43 (t,J=7 Hz, 3H); 1.79 (m,J=7.5 Hz, 2H); 2.76 (s, 3H); 2.77 (t,J=7.5 Hz, 2H); 4.45 (q,J=7 Hz, 2H); 10.44 (brs, 1H)。MS(ESI+),m/z:225.2[M+H]+。
1.2.4 化合物7a和7b的合成
将化合物6(2.24g,10mmol),无水碳酸钾(1.52g,11 mmol)和50毫升乙腈置于圆底烧瓶中,室温搅拌均匀后,加入联苯溴苄衍生物(6.1g,11mmol),所得混合物加热至回流并在该温度下反应10小时,薄层色谱检测原料完全反应,将反应液冷却至室温,过滤,滤饼用二氯甲烷洗涤数次,合并滤液,滤液浓缩至干后经柱层析分离得到化合物5.5克7a和1.04克7b。化合物7a,熔点:180~181℃,1H-NMR(CDCl3, 500 MHz)δ:7.89 (d, 1H), 7.49 (m, 2H), 7.35~7.24 (m, 10H), 7.10 (d, 2H), 6.94 (d, 6H), 6.80 (d, 2H);化合物7b, 熔点:150~152℃,1H-NMR(CDCl3, 500 MHz)δ: 7.89 (d,1H), 7.46 (m,2H), 7.35~7.25 (m, 10H), 7.09 (d, 2H), 6.94 (d, 6H), 6.73 (d, 2H), 5.23 (s, 2H), 4.43 (q, 2H), 2.60 (m, 2H), 2.38 (s, 3H), 1.70 (m, 2H), 1.41 (t, 3H), 0.91 (t, 3H)。
2 结果与讨论
目标产物的合成需要5步反应,在整个反应路线中,影响收率的关键步骤是由化合物1制备化合物3的分子间的缩合成环反应,本实验对改步反应的条件进行了优化。另外,实验重点还就如何确定化合物7a和7b的结构进行了研究。
2.1 化合物3制备过程反应条件的确定
化合物1制备化合物3的过程涉及分子间反应和分子内反应,首先,原丁酸三甲酯与二氨基马来腈中的一个氨基发生分子间反应形成中间体,所得中间体与分子结构中另外一个氨基发生分子内环合反应形成咪唑环。一般来说,为了获得满意的反应速率,分子间反应可采用高一点的底物浓度;而对于分子内的环合反应,为了避免副反应的发生,一般采用较低的底物浓度。对于化合物3的制备过程是包含分子间和分子内反应的复杂过程,因此,反应的条件对产品的收率影响较大。为讨论方便,将该反应过程分为缩合和环化两个过程,并主要考察了两个过程中溶剂对产品收率的影响,反应温度均为溶剂的沸点,以原料反应完全为反应终点。具体结果如表1所示。

表1 不同反应条件对化合物3收率的影响
由于环合过程都采用了相同的溶剂二甲苯,因此,影响产品收率的关键在于缩合过程中的溶剂。由表1中反应结果看出采用乙腈作为缩合反应溶剂时收率最高,达到了98%。如果缩合反应过程和环合反应过程采用相同的溶剂二甲苯,产品收率只有84%,与两个过程采用不同溶剂的产率差别较大。采用甲苯与二氧六环为溶剂时产品收率均大于80%,但也远远低于采用乙腈为溶剂时的收率。采用其它极性的溶剂如四氢呋喃、乙醇、甲醇时,收率均较低。因此,实验最终选择乙腈/二甲苯分别作为化合物3制备过程中的溶剂。
2.2 化合物6的结构分析
化合物6的结构中含有一个咪唑环,咪唑环本身存在共振异构,所以化合物6也存在着如图3所示的共振异构。
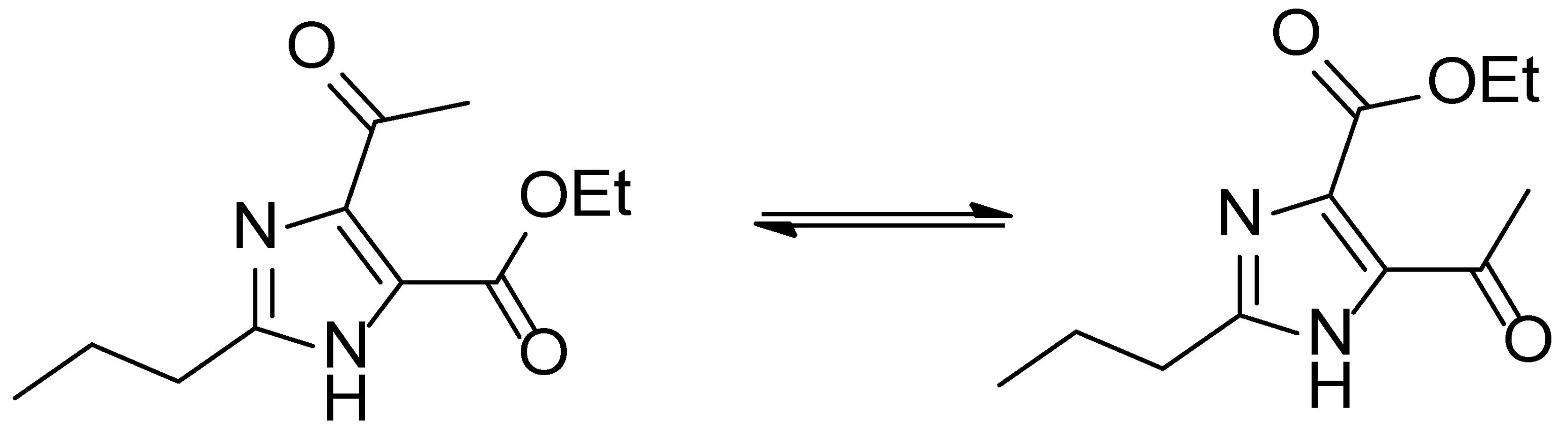
图3 咪唑环的共振异构
从化合物6的核磁图(如图4所示)可以看出,谱图中确实存在着一对异构体,通过分子结构中乙酯基团中亚甲基的峰面积比较可以得出其异构体比例为5.3∶1,但不能确认其中主要以哪种异构体存在。
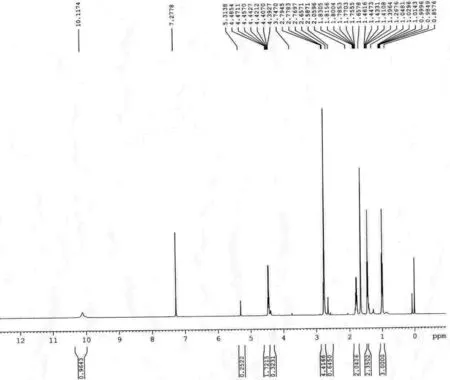
图4 化合物6的核磁氢谱
2.3 化合物7a和7b的结构确定
化合物7a和7b是一对同分异构体,由于其原料化合物6中主要异构体的构型不能确定,所以7a和7b的结构很难确定,从化合物7a和7b的核磁氢谱(如图5所示)上可以看出,芳环区氢的化学位移差别不大,乙酯基和乙酰基的化学位移差别较大,因此,可以断定联苯环与这两个基团之间在空间上有明显的效应。
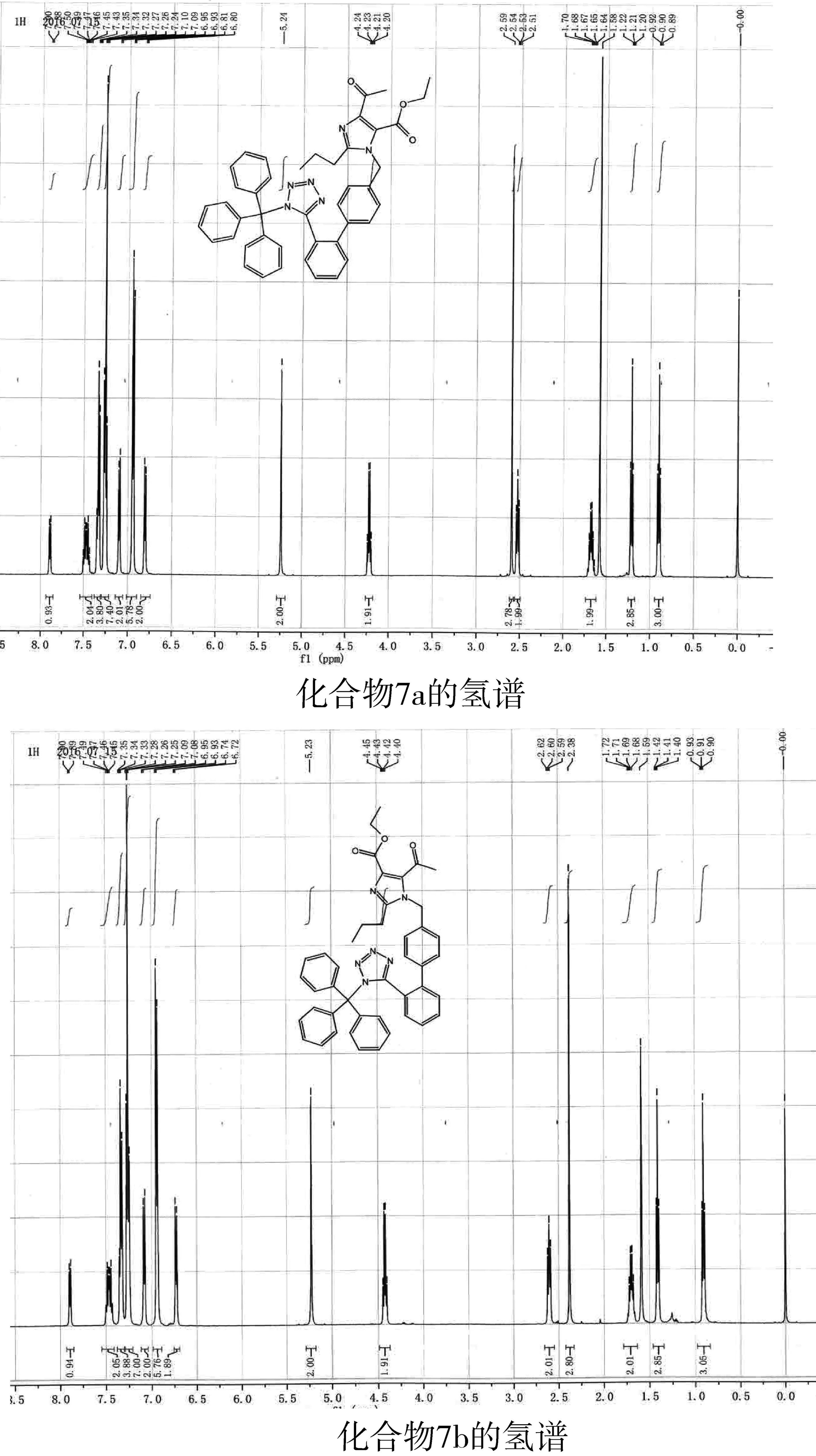
图5 化合物7a和7b的核磁共振氢谱
实验将这两个化合物分别进行了NOE研究,具体的NOE谱图如图6所示。通过照射空间位置上邻近的氢,从而确定了化合物7a和7b的结构从化合物7a的NOE图上可是看出,当照射苄位上亚甲基氢的时候,乙酯基中的亚甲基有增益,说明联苯苄基基团与酯基在空间上是邻近的,同样,当照射化合物7b上苄位上亚甲基氢的时候,乙酰基中的甲基信号有增益,说明说明联苯苄基基团与乙酰基在空间上是邻近的。由此,确认了化合物7a和7b的结构。同时也可以确定化合物6中异构体的主要构型。
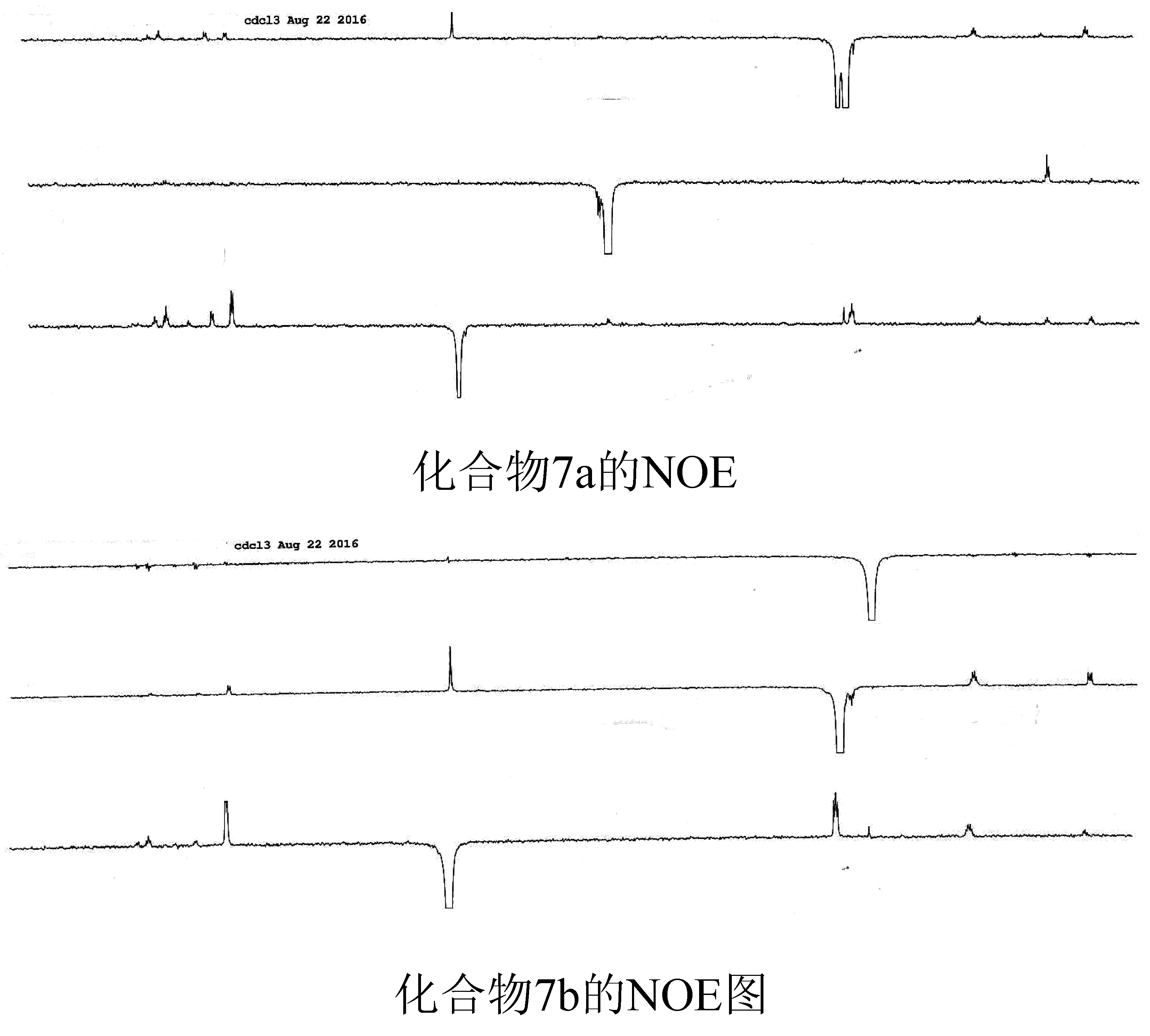
图6 化合物7a和7b的NOE谱图
3 结论
以二氨基马来腈和原丁酸三甲酯为原料,经过5步反应,合成了奥美沙坦酯中的关键中间体中的主要杂质7a,并采用核磁共振氢谱和NOE谱图确定了7a的结构,并通过HPLC分析比对,确认了奥美沙坦酯关键中间体中的主要杂质为7a,该研究结果为奥美沙坦酯质量控制提供了很好的借鉴。
[1] Tao X, Liu G L. Olmeasartan: a novel angiotensinⅡreceptor antagonist[J]. Chin J New Drugs Clin Rem, 2003, 11: 368-370.
[2] Fukuda M, Yamanaka T, Mizuno, et al. AngiotensinⅡ type 1 receptor blocker, olmesartan, restores noctumal blood pressure decline by enhancing daytime natriuresis[J]. Hypertension, 2008, 26(3): 583-588.
[3]Nobuya K, Miwa S, Shoko K, et al. An angiotensin type 1 receptor antagonist, olmesartan medoxomil, improves experimental liver fibrosis by suppression of proliferation and collagen synthesis in activated hepatic stellate cells[J]. Pharmacol.,2003, 139(6): 1085-1094.
[4]Yanagisawa H, Amemiya Y, Kanazaki T, et al. Nonpeptide Angiotensin II Receptor Antagonists: Synthesis, Biological Activities, and Structure-Activity Relationships of Imidazole-5-carboxylic Acids Bearing Alkyl, Alkenyl, and Hydroxyalkyl Substituents at the 4-Position and Their Related Compounds[J]. J Med Chem, 1996, 39: 323-338.
[5]Gardner S F, Franks A M. Olmesartan medoxomil: the seventh angiotensin receptor antagonist[J]. Annals of Pharmacother, 2003, 37(1): 99-105.
[6]Davis D P, Kirk K L, Cohen L A. New Synthesis of 2-Nitroimidazoles[J]. J Heterocycl Chem, 1982, 19: 253-256.
[7]Brunner H R, Gavras H, Laragh J H, et al. Angiotensin-II Blockade in Man by Sar1-Ala8-Angiotensin II for Understanding and Treatment of High Blood-pressure[J]. Lancet, 1973, 1045-1048.
[8]Streeten D H P, Anderson G H Jr, Dalakos T G. Angiotensin Blockade: Its Clinical Significance[J]. Am J Med, 1976, 60: 817-824.
[9]Ogihara T, Yamamoto T, Kumahara Y. Clinical Applications of Synthetic Angiotensin II Analogue[J]. Jpn Circ J, 1974, 38: 997-1003.
[10]Hata T, Ogihara T, Mikami H, Nakamaru M, et al. Comparison of the Biological Effects of Two Angiotensin II Analogues in Hypertensive Patients with Sodium Depletion[J]. Life Sci, 1978, 22: 1955-1962.
信息简讯
“医用制氧机质量分析仪的研制”通过验收
由甘肃省计量研究院承担的国家质检总局科研计划项目“医用制氧机质量分析仪的研制”,日前通过国家质检总局验收。该项目于2015年10月立项,历经一年多完成。医用制氧机质量分析仪可实现实时、在线、关键指标的检测,具有携带方便、易于操作的特点,可以现场实时对医疗机构和家用的医用制氧机进行关键参数的检测,具有很好的推广应用价值,可广泛应用于各级检验检测机构。验收委员会听取和审议了项目组的工作汇报和相关技术文件,现场审查了相关证明材料,认为该项目较好的完成了计划任务书的任务目标,各项技术指标和功能均达到项目任务书要求,部分指标优于项目任务书要求,项目成果具有良好的科学性和实用性,填补了国内空白,一致同意通过验收。
(中新网武振宁)
Study on main impurity structure of key intermediate of olmesartan.
Li Hong
( Liaoning Province Academy of Analytic Sciences,Standard System Engineering Research Center of Liaoning Province,Shenyang 110015,China)
The main impurity structure of key intermediate 8 of olmesartan was studied,and the possible structure was speculated by LC-MS. The possible structures (7a and 7b) were synthesized by the use of maleonitrile and trimethyl orthobutyrate as raw material and 5-step reaction. The structures of isomers 7a and 7b were confirmed by1H-NMR and NOE. Compariing the HPLC retention time of 7a and impurity of 8, the structure of the main impurity of key intermediate 8 was confirmed as 7a. This study provide a good reference for the quality control of olmesartan.
key intermediate of olmesartan; impurity; structure analysis
10.3969/j.issn.1001-232x.2017.04.018
2017-05-02
李红,女,1978年出生,硕士研究生,从事化工产品合成及分析研究,Email:lihong123dd@163.com。
猜你喜欢
现代实用医学(2022年10期)2022-12-08
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
国际公关(2017年4期)2017-09-18
国际公关(2016年3期)2016-06-29
国际公关(2016年1期)2016-03-01
国际公关(2016年1期)2016-03-01
合成化学(2015年2期)2016-01-17
股市动态分析(2015年12期)2015-09-10
药学与临床研究(2015年6期)2015-03-06
