
HIV-1整合酶抑制剂研究进展
2017-08-22 04:10:28朱梅,张国宁,白晓光等
中国医药生物技术 2017年4期
HIV-1整合酶抑制剂研究进展
艾滋病,又称获得性免疫缺陷综合征(acquired immune deficiency syndrome,AIDS),是由人类免疫缺陷病毒(human immunodeficiency virus,HIV)感染引起的全身性免疫系统严重损坏为特征的传染性疾病。自 1981年美国报道首例艾滋病病例以来[1],其不断向世界各地蔓延并在全球范围内肆虐流行,给人类的生命健康和社会发展造成了十分严重的危害,已引起各国政府和社会的普遍关注。我国整体上属于 HIV 低流行国家,但局部地区呈高发流行[2-3]。截至 2015年 10月底,全国报告存活的艾滋病病毒感染者和病人共57.5 万例,死亡 17.7 万人。
1 HIV 病原学和流行病学
1.1 HIV 的病原学
1983年,法国巴斯德研究所科学家 Lue Mont Lagnier等首先发现艾滋病是由一种被称为 HIV 的病毒所引起的[4-5]。次年,这一研究结果被美国科学家 Gallo 等证实[6-9]。
HIV 属于逆转录病毒科慢病毒属[10],为单股正链 RNA病毒。该病毒呈球形,直径为 100~120 nm,内含两个单链病毒 RNA 基因组和病毒逆转录酶、整合酶和蛋白酶。病毒外层囊膜系双层脂质膜,嵌有 gp120 和 gp41 两种糖蛋白。囊膜内层为 p17 蛋白构成的壳,其内还有核心蛋白 p24 包裹的 RNA,如图 1 所示[11]。
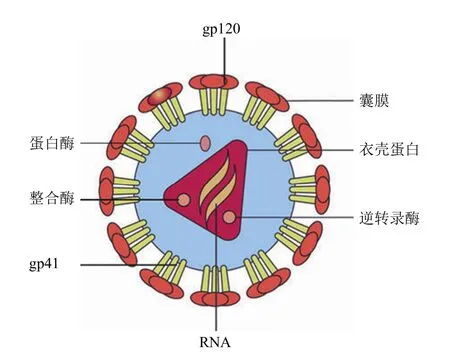
图1 HIV 病毒结构示意图
1.2 HIV 的生命周期
HIV 病毒自身不能复制,必须侵入宿主细胞后才能进行复制。HIV 在血液中的半衰期小于 6 h,但进入细胞内每天产生 1010~1012病毒颗粒,每年大约可繁殖 140 代。HIV 的复制过程分为 7 个步骤[12]:首先是病毒对宿主细胞的依附,进攻宿主细胞;HIV 选择性地感染人体免疫系统中带有 CD4 受体的细胞,如淋巴细胞、单核巨噬细胞、树突状细胞等(这是由于病毒表面糖蛋白 gpl20 和 gp4l 能特异性识别细胞表面的 CD4 受体);然后病毒表面的糖蛋白发生构象变化后与细胞膜进行融合进入细胞;单链病毒RNA 在逆转录酶作用下进行逆转录合成双链 DNA;双链病毒 DNA 在整合酶的作用下进入细胞核内,并转录病毒RNA;之后进行蛋白质的表达合成长链蛋白;最后 RNA、酶、结构蛋白在细胞内合成新病毒,成熟后溢出细胞,进而转攻其他细胞。
1.3 HIV 的流行病学特征
HIV 是目前已知分化、变异程度最高的病毒,根据血清学反应、基因序列差异和地理分布特征,可分为两个亚型:HIV-1 和 HIV-2,两者都来自非洲中西部,并从灵长类动物传播到人类。前者可能是从黑猩猩的猴免疫缺陷病毒跨种感染进化而来;后者可能是从几内亚比绍的乌黑白眉猴的另一种猴免疫缺陷病毒跨种感染而来[13]。HIV-1 是引起艾滋病全球流行的病原体,在感染人数上占 95% 的绝对优势;HIV-2 主要局限在非洲中部和西部的一些地区,感染人数不超过总数的 20%[14-15]。
自 1985年我国首次发现 HIV 感染者以来,各地开始呈现艾滋病流行态势。艾滋病在我国传染病防治法中被列为乙类传染病,属于性传播疾病。目前我国 HIV-1 主要的流行毒株之一是 HIV-1 CRF-BC 重组亚型,包含我国特有的HIV-1 毒株 CRF07-BC 和 CRF08-BC[16]。
2 HIV-1 整合酶抑制剂
2.1 整合酶的结构功能
逆转录酶(RT)、蛋白酶(PR)和整合酶(IN)是 HIV病毒基因在复制过程中的 3 个关键酶[17]。HIV-1 整合酶为病毒所特有,相对分子质量为 32 kD,由病毒的 3' 端 pol基因编码含有 288 个氨基酸的蛋白质,分为 3 个功能结构域:锌结合 N 端区域、催化核心区域和 DNA 结合 C 端区域[18]。其中 N 端区域由第 1~49 位氨基酸组成,形成HHCC 基序,能与锌离子形成锌指结构,对酶与病毒 DNA稳定起着关键作用;催化核心区域由第 50~212 位氨基酸组成,其中 Asp64、Asp116、Glu152 为酶的活性中心,形成 DDE 结构,可结合 1~2个金属阳离子;C 端区域由第 213~288 位氨基酸组成,是与 DNA 非特异性结合的区域(图 2)[19]。其中催化核心区域晶体结构已有文献报道(图 3)[20]。
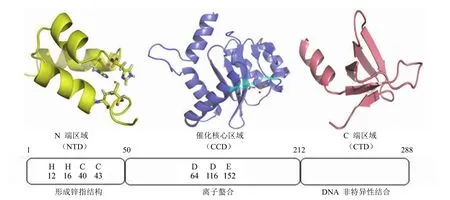
图2 整合酶功能结构域示意图
整合酶催化病毒 DNA 与宿主 DNA 的整合过程包括两个步骤:3' 端切除反应和链转移反应。整合酶与病毒双链 DNA 形成复合物,在 3' 末端各切掉两个核苷酸,暴露出 3' 末端的羟基,之后含有 HIV-1 遗传信息的双链 DNA被运送到宿主细胞核内,整合酶切割宿主细胞 DNA 的5' 端产生交错切口,将病毒 DNA 的 3' 端与宿主 DNA 的5' 端以共价键连接,由此在整合酶的作用下将病毒基因插入到宿主染色体中,完成整合过程。
2.2 HIV-1 整合酶抑制剂研究进展
由于 HIV-1 整合酶为病毒所特有,因此整合酶成为抗HIV 药物研发的理想靶点[21]。自第一个 HIV-1 整合酶抑制剂雷特格韦(1)[22]于 2007年被美国 FDA 批准上市以来,抗艾滋病药物领域有了新的突破,关于 HIV-1 整合酶的研究风起云涌。HIV-1 链转移反应抑制剂(HIV-1 INSTIs)是目前研究最多的整合酶抑制剂,埃替格韦(2)[23]、度鲁特韦(3)[24]也被 FDA 批准上市,S/GSK1265744(4)进入II 期临床研究[25](图 4)。
其他在研的 HIV-1 整合酶抑制剂根据化合物的结构可分为以下十种类型[26]:
2.2.1β-二酮酸类整合酶抑制剂β-二酮酸类化合物(图 5)具有整合酶转移反应抑制活性,且具有较强的细胞内抗病毒活性。二酮酸结构被认为是产生酶抑制活性的关键药效基团,而芳香基团主要是改善化合物的药效性质和选择性[27]。日本 Shionogi 公司合成化合物 5-CITEP(5),并得到了 HIV-1 整合酶与抑制剂复合物的 X 射线衍射晶体结构[28]。Merk 公司通过高通量筛选组合化合物库的方法得到两个小分子化合物 L-731988(6)和 L-708906(7),对链转移的 IC50分别为 80 和 150 nmol/L,且细胞内的抗 HIV 活性也很高[29]。第一个进入临床研究的 HIV 整合酶抑制剂 S-1360(8)是利用生物电子等排原理用含氧杂环取代芳香环,三氮唑基团来取代羧基得到的,其体外抑制整合酶活性的 IC50为 20 nmol/L,细胞毒性 CC50为12 μmol/L,但在 II 期临床研究中发现,S-1360 三氮唑基团在人体内的关键代谢产物不稳定而产生毒性,于 2003年终止临床试验[30]。
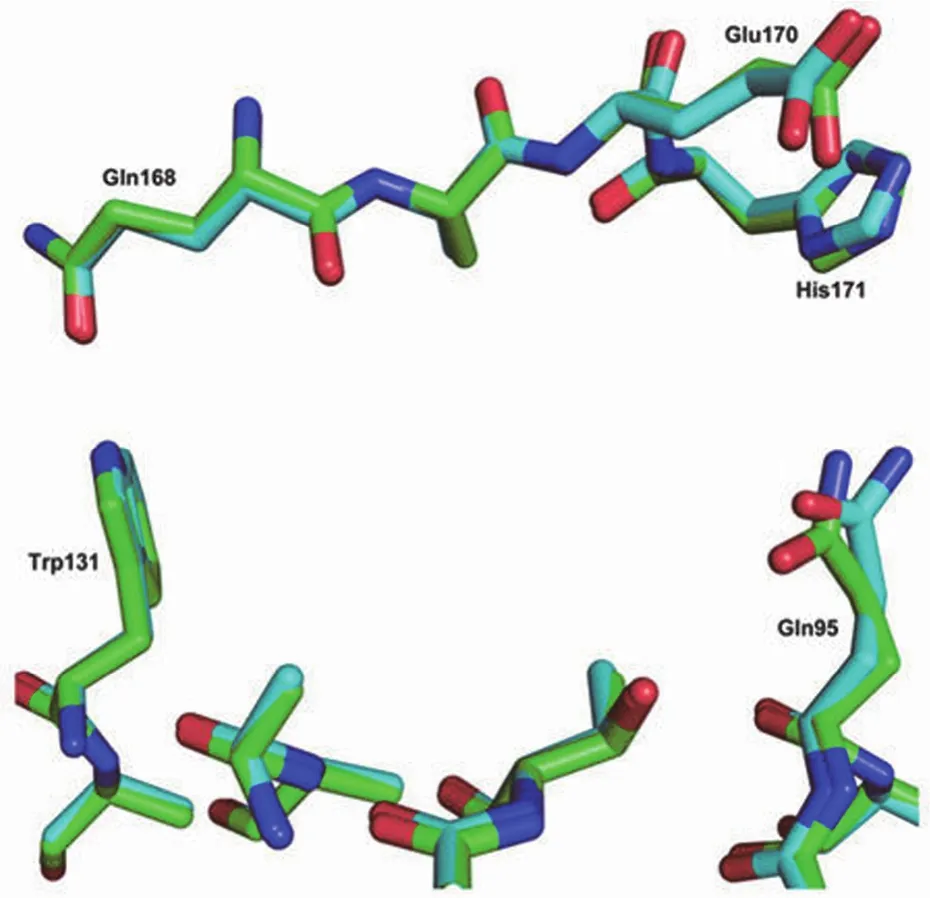
图3 整合酶核心区域晶体结构示意图
此外,武燕彬等[31]设计合成了 6 个双二酮酸类化合物,并探讨此类化合物抑制 HIV-1 整合酶活性的构效关系。双二酮酸对 HIV-1 整合酶的活性均小于 20 μg/ml,双二酮酸之间距离大则对 3' 端切除反应(3P)的抑制活性差,推断 3P 活性受两个二酮酸距离影响较大,化合物对 HIV-1整合酶的抑制活性主要表现为链转移反应(ST)的活性。
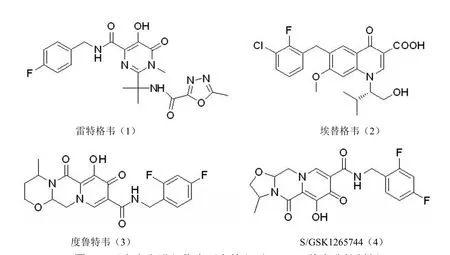
图4 已上市和进入临床研究的主要 HIV-1 整合酶抑制剂
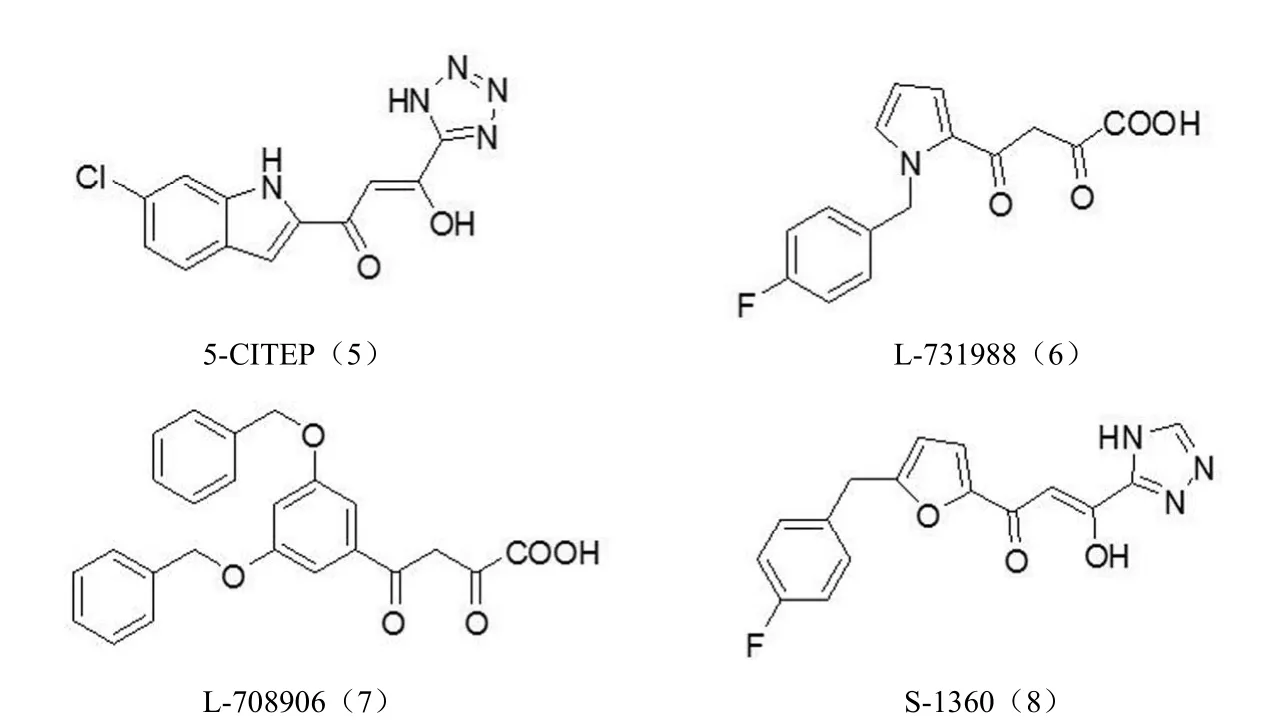
图5 β-二酮酸类整合酶抑制剂
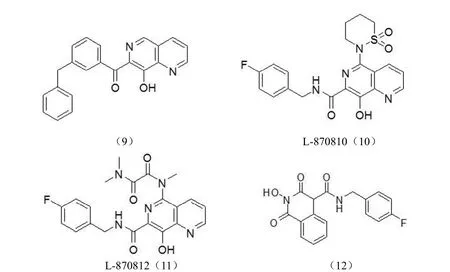
图6 1,6-二氮杂萘、喹诺酮酸及其衍生物类整合酶抑制剂
2.2.2 1,6-二氮杂萘、喹诺酮酸及其衍生物类整合酶抑制剂 由于β-二酮酸类化合物结构的不稳定性,化学家考虑将β-二酮酸用 1,6-二氮杂萘结构替代,目前已合成多个此类化合物(图 6)。这些化合物改善了抗病毒活性和药代动力学特征,提高了口服生物利用度。其中化合物(9)具有很好的抑制活性,其抑制链转移活性 IC50值为 0.01 μmol/L,抗病毒活性 IC95为(0.39 ± 0.16)μmol/L。化合物 L-870810(10)和 L-870812(11)对链转移的 IC50分别为 15 和40 nmol/L[27],但长期大剂量使用易产生肝毒性和肾毒性。
根据电子等排体原理,Sato等[32]将β-二酮酸结构替换为喹诺酮酸结构,其 IC50值为 7.2 nmol/L,既保持了很好的活性,代谢过程又优于β-二酮酸类整合酶抑制剂,目前埃替格韦(2)已被 FDA 批准上市。
通过分析埃替格韦等喹诺酮酸类构效关系,Billamboz等[33]合成了一系列 2-羟基-1,3-二氧代-2H,4H-异喹啉-4-酰胺类化合物,其中化合物(12)同时具有 3' 端切除反应和链转移反应抑制活性,其 IC50和 EC50分别为 0.056 μmol/L和 2.34 μmol/L,有望成为新一代的 HIV-1 整合酶抑制剂。
2.2.3 嘧啶酮类整合酶抑制剂 2007年被美国 FDA 批准的雷特格韦(1)是首个上市的 HIV-1 整合酶抑制剂,属于嘧啶酮类。它具有良好的理化性质和药代动力学性质,IC50浓度在纳摩尔级,可以与其他抗艾滋病药物合用[22],开辟了抗艾滋病药物研究的新领域。Merck 公司以嘧啶二酮化合物为先导化合物进行结构修饰,得到了抗 HIV 活性增强的一些化合物,其中化合物(13)在 50% 人类血清中IC95值为 10 nmol/L[34]。Gu 等[35]设计合成了一系列嘧啶酮类化合物,其中化合物(14)和(15)对野生型 HIV-1病毒具有很好的抑制活性,EC50值分别为 2.6 nmol/L 和3.0 nmol/L,且选择指数均大于 1000(图 7)。
2.2.4 硫氮硫扎平类整合酶抑制剂 Neamati 等[36]合成了一系列硫氮硫扎平类整合酶抑制剂(图 8),此类化合物作用于 HIV-1 整合酶的链转移反应步骤,具有一定的抑制活性。构效关系研究表明,萘环的引入能够提高化合物的抑制活性,意味着蛋白酶的活性疏水口袋可能与芳香体系相匹配。氢键的引入对化合物的活性没有明显的改变作用。化合物(16)、(17)、(18)的 IC50值分别为(58 ± 15)、52 和(40 ± 10)μmol/L。
2.2.5 多羟基整合酶抑制剂 研究显示,黄酮及黄酮烷类等多羟基化合物具有 HIV-1 整合酶抑制活性,当羟基被取代时,活性消失。Mekouar 等[37]根据这一思路设计合成了一系列苯乙烯基喹啉类化合物(SQLs)。这类化合物作用于HIV-1 整合酶的催化核心区域,与金属阳离子产生螯合作用,显示了极强的体外 HIV-1 整合酶抑制活性,且在浓度达到 100 μmol/L 时也无细胞毒性,一些化合物的 IC50值甚至达到了亚微摩尔级,其中喹啉环 7 位游离羟基是保持活性的必需基团。
此外,Yang 等[38]还合成了一类哌嗪类多羟基化合物(图 9),均显示了一定的抑制活性,此类化合物作用于整合酶的链转移反应阶段,研究显示与酰胺键相连的芳环上有三羟基取代的化合物具有良好的抑制活性,而二羟基取代化合物几乎无抑制活性,如化合物(19)的 IC50为12.8 μmol/L,而化合物(20)的 IC50> 100 μmol/L。
2.2.6 偕二砜类整合酶抑制剂 根据菊苣酸(21)的结构,Meadows 等[39-40]设计了一类电中性的偕二砜类化合物(图 10),大部分显示了良好的 HIV-1 整合酶抑制活性。研究结果显示,吸电子取代基会导致抗病毒活性和细胞毒性同时提高,给电子取代基能够显著降低细胞毒性,但同时也失去抗病毒活性。其中化合物(22)的 IC50值为(33 ±3)μmol/L,而化合物(23)的 IC50> 100 μmol/L。
2.2.7 苯磺酰胺类整合酶抑制剂 Nicklaus 等[41]根据具有 HIV-1 整合酶抑制活性的咖啡酸苯乙基酯(24)设计了虚拟的三点药效团模型,根据该药效团模型筛选出 267 个化合物,对其中 60 个化合物进行体外 HIV-1 蛋白酶抑制活性的筛选,发现有 19 个化合物在微摩尔浓度水平下具有同时抑制 3' 端切除反应和链转移反应的活性,其中含有苯磺酰胺结构的化合物,如化合物(25)具有显著的 HIV-1 整合酶抑制活性(图 11)。
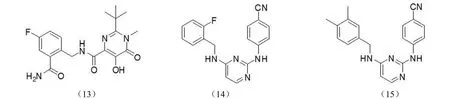
图7 嘧啶酮类整合酶抑制剂
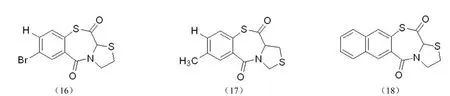
图8 硫氮硫扎平类整合酶抑制剂
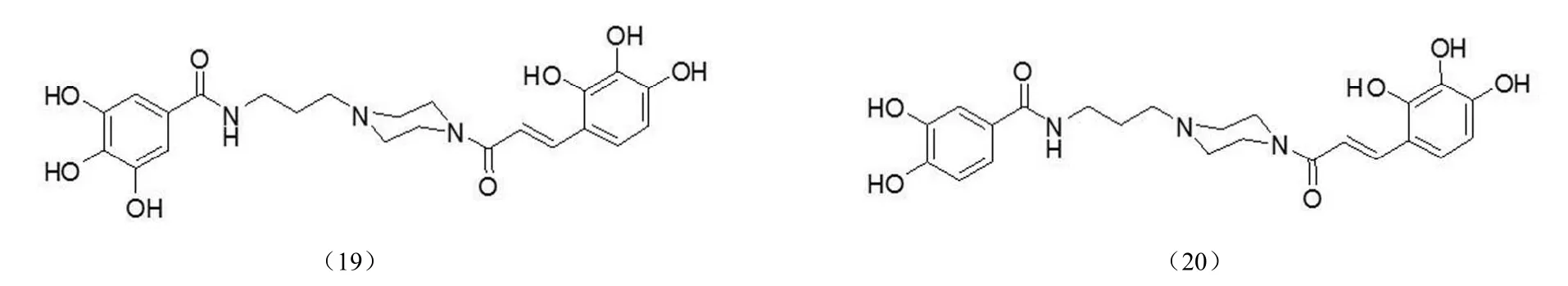
图9 多羟基整合酶抑制剂
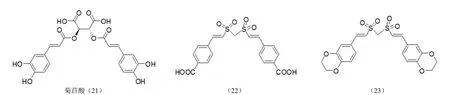
图10 偕二砜类整合酶抑制剂
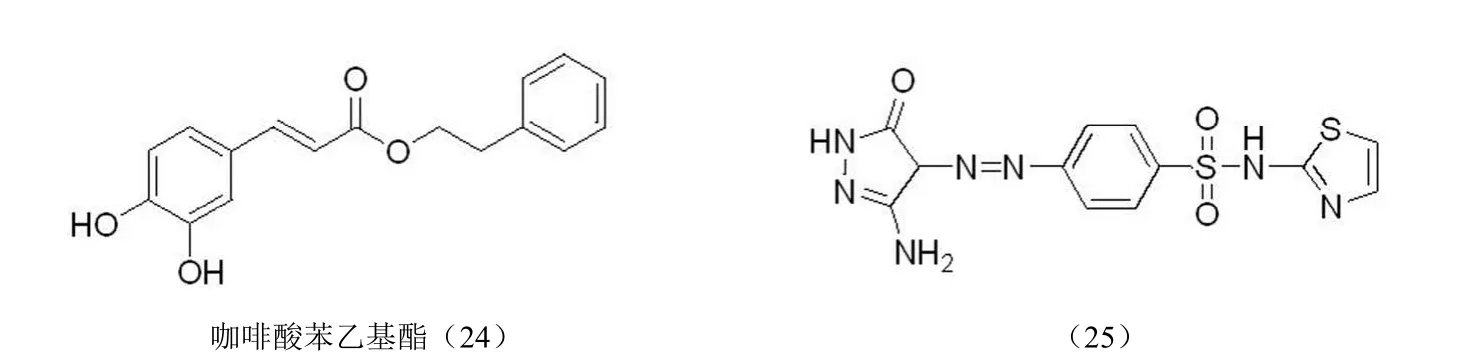
图11 苯磺酰胺类整合酶抑制剂
2.2.8 香豆素整合酶抑制剂 Mazumder 等[42]研究发现,含有 4 个 4-羟基香豆素片段的化合物 NSC158393(26)(图 12)具有抗病毒和抑制 HIV-1 整合酶的性质。在微摩尔浓度水平时具有 3' 端切除的抑制活性和抑制链转移反应的活性,此外还发现该类化合物具有 HIV-1 蛋白酶抑制活性。通过评价 NSC158393 及其类似物发现羟基和羰基是药效团的必需结构。羟基香豆素有望发展成为同时具有HIV-1 整合酶和蛋白酶抑制活性的一类新型先导化合物。
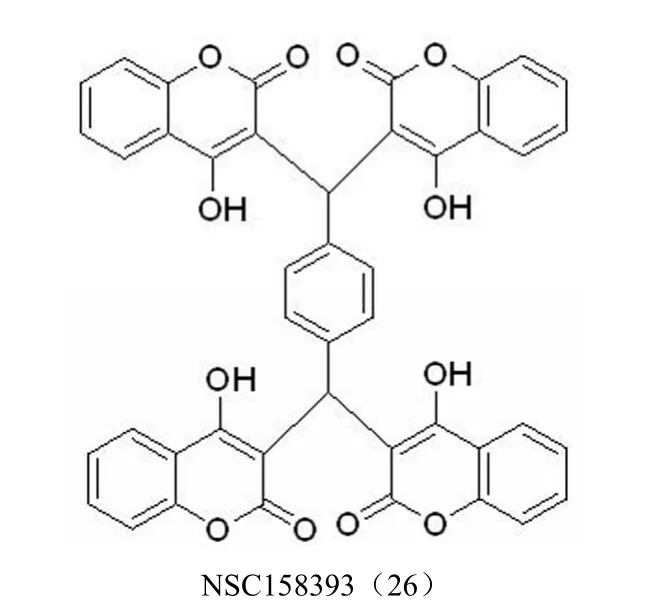
图12 香豆素整合酶抑制剂
2.2.9 水杨酰肼类整合酶抑制剂 化学家发现了一类具有HIV-1 整合酶抑制活性的水杨酰肼类化合物[43],并设计合成了一系列结构化合物(图 13),其中化合物(27)、(28)、(29)、(30)的 IC50值分别为 8.2、4.4、0.053、0.062 μmol/L。研究发现,水杨基片段上的羟基是活性必需基团,当用氨基、卤素、羧基、醚等取代时将导致活性完全丧失;此外对分子结构中其他片段的修饰也会影响化合物的活性。
2.2.10 其他类型整合酶抑制剂 de Soultrait 等[44]从真菌发酵液中分离得到由芳香氨基酸组成的双环六肽化合物Complestatin(31、32)同时具有 3' 端切除和链转移抑制活性。Hadi 等[45]合成了一系列吡唑啉酮结构化合物,其中化合物(33)具有良好的抑制整合酶链转移反应的活性,其IC50为(3 ± 0.4)μmol/L,为新型 HIV-1 整合酶抑制剂的研发提供了丰富的信息和思路(图 14)。
2.3 整合酶/逆转录酶双靶点抑制剂
近年来,系统生物学和网络药理学的发展使多靶点药物设计成为当前研究的前沿领域,并设计合成了一系列整合酶/逆转录酶(IN/RT)双靶点 HIV-1 抑制剂[46](图 15)。
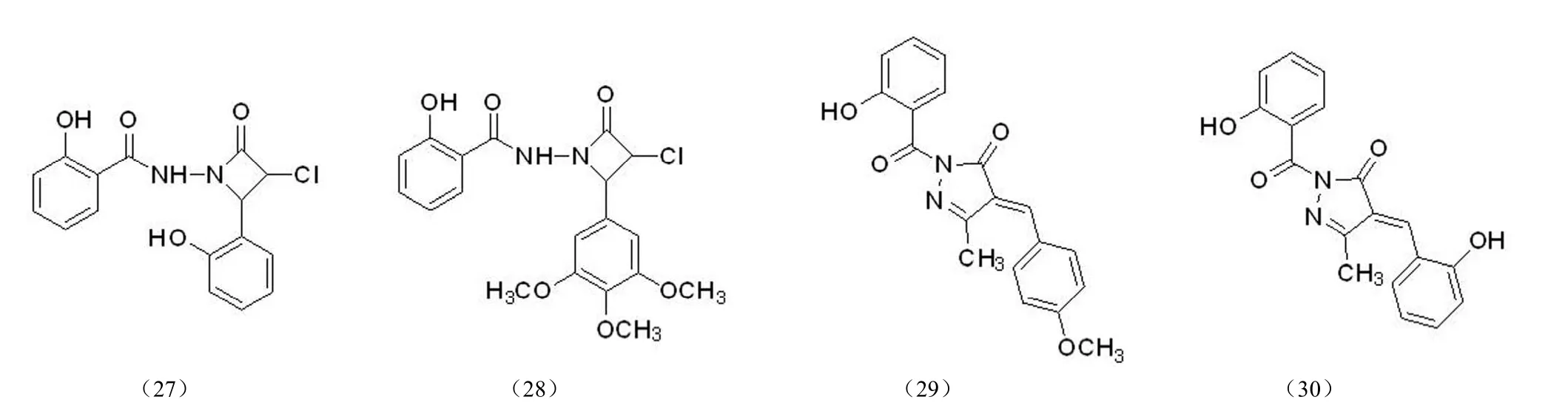
图13 水杨酰肼类整合酶抑制剂
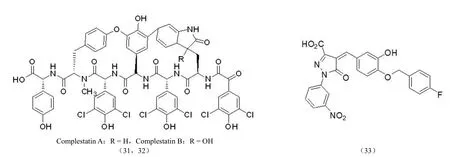
图14 其他类型整合酶抑制剂
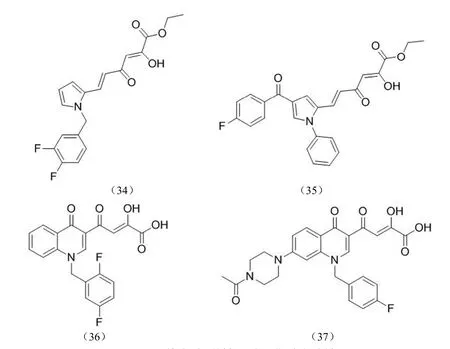
图15 整合酶/逆转录酶双靶点抑制剂
HIV-1 整合酶和逆转录酶核糖核酸 H 域(RNase H)均是 HIV 治疗的靶点,据此,Cuzzucoli Crucitti 等[47]设计合成了一系列吡咯基-β-二酮酸结构化合物,部分化合物对两种酶均有抑制作用,其中化合物(34)、(35)对 IN 的IC50分别为 3.0 和 1.2 μmol/L;对 RNase H 的 IC50分别为 0.6 和 1.8 μmol/L。
7 位胺基取代的喹诺酮-β-二酮酸类化合物不仅可以抑制整合酶链转移反应,同时能够抑制 RNase H,同一课题组 Pescatori 等[48]设计合成了一系列化合物,在喹诺酮的7 位引入烷基取代的胺基基团,在 1 位引入取代苯基,其中活性最好的化合物(36)对蛋白酶的 IC50为 10 nmol/L,仅有部分化合物对 RNase H 有抑制活性,如化合物(37)对 RNase H 的 IC50为 3.3 μmol/L,对 IN 的 IC50为80 nmol/L。
3 展望
随着研究的不断发展和深入,抗 HIV 药物呈现出多样化,为艾滋病的治疗提供了可靠的保障。但是由于耐药株的出现,为 HIV 的治疗带来了挑战,开发出生物利用度高、活性好、抗耐药性的药物成为战胜艾滋病的关键。目前耐药性的研究主要集中于所设计的化合物能与酶产生分子间作用力;此外,植物、微生物和海洋生物等来源的新型天然药物也逐渐成为研究热点。针对 HIV-1 整合酶抑制剂生物利用度低的问题,通常与药效增强剂合用;在抑制剂分子结构中引入靶向片段或基团以提高其生物利用度也是一种较为有效的手段。此外,组合化学、基因学、计算机辅助药物设计、合成生物技术等多学科的发展和综合利用,将为抗 HIV药物的研发提供更广阔的前景。
[1]Centers for Disease Control (CDC).Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men--New York City and California.MMWR Morb Mortal Wkly Rep, 1981, 30(25):305-308.
[2]Gao YJ, Liu YJ, Jiang SL.Research status of cohort of high risk groups of HIV in China.Chronic Pathemathol J, 2013, 14(3):231-236.(in Chinese)
高彦杰, 刘英杰, 姜树林.国内HIV高危人群队列研究现状.慢性病学杂志, 2013, 14(3):231-236.
[3]Xu P, Han L, Zeng G, et al.Prevention and control of sexuallytransmitted HIV/AIDS in China: Policy evolution and trends.Chin J Health Policy, 2013, 6(7):64-70.(in Chinese)
徐鹏, 韩琳, 曾刚, 等.我国预防控制经性途径传播艾滋病的政策变迁及趋势分析.中国卫生政策研究, 2013, 6(7):64-70.
[4]Barré-Sinoussi F, Cherman JC, Rey F, et al.Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS).Science, 1983, 220(4599):868-871.
[5]Montagnier L.Historical essay.A history of HIV discovery.Science,2002, 298(5599):1727-1728.
[6]Popovic M, Sarngadharan MG, Read E, et al.Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS.Science, 1984, 224(4648):497-500.
[7]Gallo RC, Salahuddin SZ, Popovic M, et al.Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS.Science, 1984, 224(4648):500-503.
[8]Schüpbach J, Popovic M, Gilden RV, et al.Serological analysis of a subgroup of human T-iymphotropic retroviruses (HTLV-III) associated with AIDS.Science, 1984, 224(4648):503-505.
[9]Sarngadharan MG, Popovic M, Burch L, et al.Antibodies reaction with human T-iymphotropic retroviruses (HTLV-III) in the serum of patients witn AIDS.Science, 1984, 224(4648):506-508.
[10]Scozzafava A, Mastrolorenzo A, Supuran CT.Carbonic anhydrase inhibitors and activators and their use in therapy.Expert Opin Ther Patents, 2006, 16(12):1627-1664.
[11]Mehellou Y, De Clercq E.Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J Med Chem,2010, 53(2):521-538.
[12]Reeves JD, Piefer AJ.Emerging drug targets for antiretroviral therapy.Drugs, 2005, 65(13):1747-1766.
[13]Sharp PM, Hahn BH.Origins of HIV and the AIDS pandemic.Cold Spring Harb Perspect Med, 2011, 1(1):a006841.
[14]Taylor BS, Sobieszczyk ME, McCutchan FE, et al.The challenge of HIV-1 subtype diversity.N Engl J Med, 2008, 358(15):1590-1602.
[15]Santos AF, Soares MA.HIV genetic diversity and drug resistance.Viruses, 2010, 2(2):503-531.
[16]He X, Xing H, Ruan Y, et al.A comprehensive mapping of HIV-1 genotypes in various risk groups and regions across China based on a nationwide molecular epidemiologic survey.PLoS One, 2012, 7(10):e47289.
[17]Demulemeester J, Christ F, Maeyer M, et al.Fueling HIV-1 integrase drug design with structural insights.Drug Discov Today Technol,2012, 9(3):e205-e212.
[18]Pommer Y, Marchand C, Neamati N.Retroviral integrase inhibitors year 2000: update and perspectives.Antivir Res, 2000, 47(3):139-148.
[19]Hajimahdi Z, Zarghi A.Progress in HIV-1 integrase inhibitors: a review of their chemical structure diversity.Iran J Pharm Res, 2016,15(4):595-628.
[20]Peat TS, Rhodes DI, Vandegraaff N, et al.Small molecule inhibitors of the LEDGF site of human immunodeficiency virus integrase identified by fragment screening and structure based design.PLoS One, 2012, 7(7):e40147.
[21]Zhao XZ, Smith SJ, Métifiot M, et al.4-amino-1-hydroxy-2-oxo-1,8-naphthyridine-containing compounds having high potency against raltegravir-resistant, integrase mutants of HIV-1.J Med Chem, 2014,57(12):5190-5202.
[22]Summa V, Petrocchi A, Bonelli F, et al.Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection.J Med Chem, 2008, 51(18):5843-5855.
[23]Serrao E, Odde S, Ramkumar K, et al.Raltegravir, elvitegravir, and metoogravir: the birth of "me-too" HIV-1 integrase inhibitors.Retrovirology, 2009, 6:25.
[24]Hare S, Smith S, Métifiot M, et al.Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572).Mol Pharmacol, 2011, 80(4):565-572.
[25]Ford SL, Gould E, Chen S, et al.Effects of etravirine on the pharmacokinetics of the integrase inhibitor S/GSK1265744.Antimicrob Agents Chmother, 2013, 57(1):277-280.
[26]Li Y, Xuan S, Feng Y, et al.Targeting HIV-1 integrase with strand transfer inhibitors.Drug Discov Today Technol, 2015, 20(4):435-449.
[27]Zhao G, Wang C, Liu C, et al.New developments in diketo-containing inhibitors of HIV-1 integrase.Mini Rev Med Chem, 2007, 7(7):707-725.
[28]Goldgur Y, Craigie R, Cohen GH, et al.Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design.Proc Natl Acad Sci U S A, 1999, 96(23):13040-13043.
[29]Hazuda DJ, Felock P, Witmer M, et al.Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells.Science,2000, 287(5435):646-650.
[30]Cotelle P.Patented HIV-1 integrase inhibitors (1998-2005).Recent Pat Antiinfect Drug Discov, 2006, 1(1):1-15.
[31]Wu YB, Jin J, Wang RZ, et al.Anti HIV-1 integrase activity of the compounds containing bis-diketo acid.Chin Med Biotechnol, 2015,10(5):411-415.(in Chinese)
武燕彬, 金洁, 王瑞贞, 等.双二酮酸结构 HIV-1整合酶抑制剂的合成及活性研究.中国医药生物技术, 2015, 10(5):411-415.
[32]Sato M, Motomura T, Aramaki H, et al.Novel HIV-1 integrase inhibitors derived from quinolone antibiotics.J Med Chem, 2006,49(5):1506-1508.
[33]Billamboz M, Suchaud V, Bailly F, et al.4-substituted 2-hydroxyisoquinoline-1,3(2H,4H)-diones as a novel class of HIV-1 integrase inhibitors.ACS Med Chem Lett, 2013, 4(7):606-611.
[34]Di Franceesco ME, Pace P, Fiore F, et al.Development of 2-t butyl-N-methyl pyrimidones as potent inhibitors of HIV integrase.Bioorg Med Chem Lett, 2008, 18(8):2709-2713.
[35]Gu SX, Qiao H, Zhu YY, et al.A novel family of diarylpyrimidines(DAPYs) featuring a diatomic linker: Design, synthesis and anti-HIV activities.Bioorg Med Chem, 2015, 23(20):6587-6593.
[36]Neamati N, Turpi JA, Winslow HE, et al.Thiazolothiazepine inhibitors of HIV-1 integrase.J Med Chem, 1999, 42(17):3334-3341.
[37]Mekouar K, Mouscadet JF, Dwsmaele D, et al.Styrylquinoline derivatives: a new class of potent HIV-1 integrase inhibitors that block HIV-1 replication in CEM cells.J Med Chem, 1998, 41(15):2846-2857.
[38]Yang L, Xu X, Huang Y, et al.Synthesis of polyhydroxylated aromatics having amidation of piperazine nitrogen as HIV-1 integrase inhibitor.Bioorg Med Chem Lett, 2010, 20(18):5469-5471.
[39]Meadows DC, Mathews TB, North TW, et al.Synthesis and biological evaluation of germinal disulfones as HIV-1 integrase inhibitors.J Med Chem, 2005, 48(14):4526-4534.
[40]Meadows DC, Sanchez T, Neamati N, et al.Ring substituent effects on biological activity of vinyl sulfones as inhibitors of HIV-1.Bioorg Med Chem, 2007, 15(2):1127-1137.
[41]Nicklaus MC, Neamati N, Hong H, et al.HIV-1 integrase pharmacophore: discovery of inhibitors through three-dimensional database searching.J Med Chem, 1997, 40(6):920-929.
[42]Mazumder A, Wang S, Neamati N, et al.Antiretroviral agents as inhibitors of both human immunodeficiency virus type 1 integrase and protease.J Med Chem, 1996, 39(13):2472-2481.
[43]Neamati N, Hong H, Owen JM, et al.Salicylhydrazine-containing inhibitors of HIV-1 integrase: implication for a selective chelation in the integrase active site.J Med Chem, 1998, 41(17):3202-3209.
[44]de Soultrait VR, Desjobert C, Tarrago-Litvak L.Peptides as new inhibitors of HIV-1 reverse transcriptase and integrase.Curr Med Chem, 2003, 10(18):1765-1778.
[45]Hadi V, Koh YH, Sanchez TW, et al.Development of the next generation of HIV-1 integrase inhibitors: pyrazolone as a novel inhibitor scaffold.Bioorg Med Chem Lett, 2010, 20(22):6854-6857.
[46]Zhan P, Li X, Kang DW, et al.Novel strategies in rational design of anti-HIV agents: multitarget and multivalency ligands.Chin J Med Chem, 2013, 23(5):406-416.(in Chinese)
展鹏, 李潇, 康东伟, 等.抗艾滋病药物设计新策略: 多靶点及多价态结合配体.中国药物化学杂志, 2013, 23(5):406-416.
[47]Cuzzucoli Crucitti G, Métifiot M, Pescatori L, et al.Structure-activity relationship of pyrrolyl diketo acid derivatives as dual inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease H domain.J Med Chem, 2015, 58(4):1915-1928.
[48]Pescatori L, Métifiot M, Chung S, et al.N-substituted quinolinonyl diketo acid derivatives as HIV Integrase strand transfer inhibitors and their activity against RNase H function of reverse transcriptase.J Med Chem, 2015, 58(11):4610-4623.
卫生计生委成立消除疟疾技术和重症疟疾救治专家组
为保障《全国消除疟疾工作方案(2016 – 2020年)》(国卫办疾控函〔2016〕931 号)实施,落实《国家卫生计生委办公厅关于进一步加强消除疟疾工作的通知》(国卫办疾控函〔2017〕392 号)要求,加强对各地疟疾防治和诊疗的技术支持与帮助,确保 2020年我国如期实现全国消除疟疾目标,卫生计生委决定成立国家消除疟疾技术专家组,承担输入性病例甄别审核、指导本地病例调查处置、参加消除疟疾考核评估等工作;成立国家重症疟疾救治专家组,承担重症病例规范治疗指导和规范用药检查等工作。专家组成员要认真履职,努力扎实工作,积极为全国消除疟疾提供技术指导和支持。请有关单位对专家开展工作给予大力支持和必要保障。
专家组人员名单及详情请登录国家卫生和计划生育委员会网站 http://www.nhfpc.gov.cn/xxgk/pages/viewdocument.jsp?dispatchDate=&staticUrl=%2Fjkj%2Fs5873%2F201707%2F403d8b279b874432b5a0d920ff4c620e.shtml 查阅。
Regenerative Medicine Cross Road 活动将在东京举办
日本再生医学论坛(FIRM协会)与中国医药生物技术协会系友好协会。FIRM 协会将于 2017年 9月 1日在东京举办“Regenerative Medicine Cross Road”活动,该活动每三个月举办一次。参加人员有日本制药界、FIRM会员相关公司和来自海外的学术与企业人士;活动目的为推介各自产品和技术,寻找商业合作者。通过参加活动,可洽谈诸多成果、增加合作机会和扩大中小公司业务。
FIRM欢迎有意与日本公司洽谈合作业务、寻找商业合作伙伴、推介产品和技术以及为项目征集资金的公司或人士参加本次活动。该活动无注册费,食宿交通等费用自理。请关注 https://firm.or.jp/ 网站,获悉后续活动通知。如有其他事宜也可致电:010-62115986、010-62126275 转 608,联系人:盖瀛。
朱梅,张国宁,白晓光,王菊仙,王玉成
10.3969/j.issn.1673-713X.2017.04.009
国家自然科学基金(81473099)
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所化学合成室
王玉成,Email:wyc9999@126.com
2017-05-08
猜你喜欢
煤炭与化工(2021年6期)2021-08-06 10:04:10
中华养生保健(2020年2期)2020-11-16 00:49:20
云南医药(2020年5期)2020-10-27 01:37:50
医学新知(2019年4期)2020-01-02 11:04:02
中成药(2018年2期)2018-05-09 07:20:05
新乡学院学报(2016年6期)2016-12-01 05:21:38
安徽大学学报(自然科学版)(2016年2期)2016-09-20 12:09:27
当代化工研究(2016年9期)2016-03-20 16:22:11
云南中医学院学报(2014年2期)2014-11-07 02:48:12
有机氟工业(2014年4期)2014-06-01 12:30:37
