
液相色谱-串联质谱法测定血清和尿液中硝基咪唑类药物残留
2017-08-10 10:28张金玲郭礼强吴翠玲
动物医学进展 2017年7期
张金玲,郭礼强,孙 军,吴翠玲
(1.潍坊出入境检验检疫局,山东潍坊 261041;2.安捷伦科技(中国)有限公司,北京 100102)
液相色谱-串联质谱法测定血清和尿液中硝基咪唑类药物残留
张金玲1,郭礼强1,孙 军1,吴翠玲2
(1.潍坊出入境检验检疫局,山东潍坊 261041;2.安捷伦科技(中国)有限公司,北京 100102)
为建立血清和尿液中甲硝唑、羟基甲硝唑、二甲硝咪唑和羟基二甲硝咪唑的高效液相色谱-串联质谱(HPLC-MS/MS)快速测定方法,样品用乙酸乙酯提取,经PCX固相萃取柱净化,Eclipse Plus C18色谱柱(3.0 mm×100 mm,1.8 μm)分离,进行HPLC-MS/MS分析。结果表明,甲硝唑、羟基甲硝唑、二甲硝咪唑及羟基二甲硝咪唑在0.20 μg/L~50 μg/L范围呈良好线性,线性相关系数均大于0.995,检出限为0.20 μg/L,定量下限为0.50 μg/L。样品在定量下限1、2、10倍3个加标水平下的平均回收率为89.3%~108.2%,相对标准偏差(RSD)为2.63%~10.0%。该方法灵敏、简便、准确,可用于血清和尿液中硝基咪唑类药物的检测分析。
液相色谱-串联质谱;血清;尿液;残留分析;硝基咪唑
硝基咪唑类药物(nitromidazoles)是一类应用广泛的人工合成的具有5-硝基咪唑基本结构的抗菌、抗原虫药物,甲硝唑(metronidazole)和二甲硝咪唑(dimetridazole)为硝基咪唑类常用的两种,被广泛应用于杀灭及预防厌氧菌和病原虫,也用作生长促进剂来促进鸡、猪、牛等的生长,其在动物体内迅速被代谢成羟基甲硝唑和羟基二甲硝咪唑。然而,硝基咪唑类药物具有致突变和致癌性[1],其代谢物因同样含有咪唑环而具有与原药类似的毒性[2]。欧盟在96/23/EC欧盟理事会指令及EC2377/90[3]中将其列入A类禁用药物,不得在任何食品中检出。为防止二甲硝咪唑等致癌药物在食源性动物残留对人体健康产生危害,我国也于2002年禁用了该类药物[4]。由于经济利益驱动和监管手段的不足,违法企业常在动物饲料和动物疫病防治过程中使用硝基咪唑类药物中的甲硝唑、地美硝唑(二甲硝咪唑)及其盐、酯及制剂。因此,建立血清和尿液中甲硝唑和二甲硝咪唑及代谢物残留量的分析方法,可以在屠宰前预知动物源食品风险,增加了检验检疫行政执法的手段。
目前,硝基咪唑类化合物的检测方法有气相色谱-质谱法[5]、液相色谱法[6-7]及液相色谱-串联质谱法[8-13],气相色谱-质谱法的缺点是需要衍生化,液相色谱法的选择性和灵敏度较差,具有高特异性和灵敏度的液相色谱-质谱法是目前检测硝基咪唑类药物残留的首选方法。本文应用液相色谱-串联质谱法同时检测血清和尿液中甲硝唑和二甲硝咪唑及代谢物药物残留,该方法具有快速、高效、高灵敏度等特点,为政府监管部门提供了技术支持。
1 材料与方法
1.1 材料
1.1.1 试剂 甲硝唑、羟基甲硝唑、二甲硝咪唑、羟基二甲硝咪唑、氘代二甲硝咪唑及氘代羟基二甲硝咪唑标准品,德国Dr.Ehrenstorfer公司产品;甲醇、乙酸乙酯、乙腈及甲酸均为色谱纯,美国Biopure公司产品; 盐酸和氨水为分析纯,天津科密欧公司产品。
1.1.2 溶液配制 标准溶液:分别配制甲硝唑、羟基甲硝唑、二甲硝咪唑、羟基二甲硝咪唑、氘代二甲硝咪唑及氘代羟基二甲硝咪唑终浓度均为100 ng/mL的标准溶液,于0~4℃冰箱中保存。定容液:取80 mL 1 mL/L的甲酸水(V/V),与20 mL乙腈混匀。
1.1.3 仪器设备 高效液相色谱-高分辨质谱联用仪(Agilent 1260-6460),美国Agilent公司产品;配电喷雾离子源(ESI)、离心机(5810R型),Eppendorf公司产品;氮吹仪(N-EVAP),Organomation Associates公司产品;纯水机(Milli-Q),美国Millipore公司产品;万分之一天平(Mettler AE163);混合型阳离子交换柱(200 mg/6 mL),Agilent公司产品。
1.2 方法
1.2.1 样品处理
1.2.1.1 血清 准确量取2 mL样品,置于50 mL聚丙烯离心管中,加入内标工作溶液40 μL,混匀后加入乙腈2 mL,涡混均匀,加入15 mL乙酸乙酯,2.0 g无水硫酸镁,涡混均匀,10 000 r/min离心10 min,将上清液转入另一50 mL聚丙烯离心管,残渣再次用15 mL乙酸乙酯涡混重复提取,合并提取液,氮气吹干。
1.2.1.2 尿液 准确量取4 mL样品,置于50 mL聚丙烯离心管中,加入4 mL水,再加入内标工作溶液40 μL,混匀后加入15 mL乙酸乙酯涡混均匀,10 000 r/min离心10 min,将上清液转入另一50 mL聚丙烯离心管,残渣再次用15 mL乙酸乙酯涡混重复提取,合并提取液,氮气吹干。
1.2.2 净化 用0.1 mol/L盐酸25 mL溶解样品,滤纸过滤。依次用3 mL甲醇、3 mL 0.1 mol/L盐酸活化阳离子交换柱,将上述溶解液全部过柱,依次用5 mL 0.1 mol/L盐酸,5 mL甲醇淋洗, 5 mL洗脱溶液洗脱并收集,在40 ℃水浴上氮气吹干,用1 mL初始流动相溶液涡旋溶解,过0.22 μm滤膜,供质谱测定。
1.2.3 液相色谱串联质谱条件
1.2.3.1 液相色谱条件 色谱柱:Eclipse Plus C18(3.0 mm×100 mm,1.8 μm);流动相A:甲酸水(1∶999,V/V)溶液;流动相B:乙腈;梯度洗脱程序:0 min 20%流动相B;3.0 min 40%流动相B;4.0 min 90%流动相B;4.1 min 20%流动相B;6.0 min 20%流动相B。进样体积:20μL。柱温:40 ℃;流速:0.4 mL/min。
1.2.3.2 质谱条件 大气压电喷雾电离源(ESI),正离子电离模式;干燥气:10 L/min;干燥温度:350 ℃;喷雾气:45 psi ;电子倍增器电压:400 V;毛细管电压:4 000 V;雾化气压力:275.8 Kpa(40 psi);多反应监测模式(MRM);甲硝唑和二甲硝咪唑及代谢物的部分质谱参数见表1。
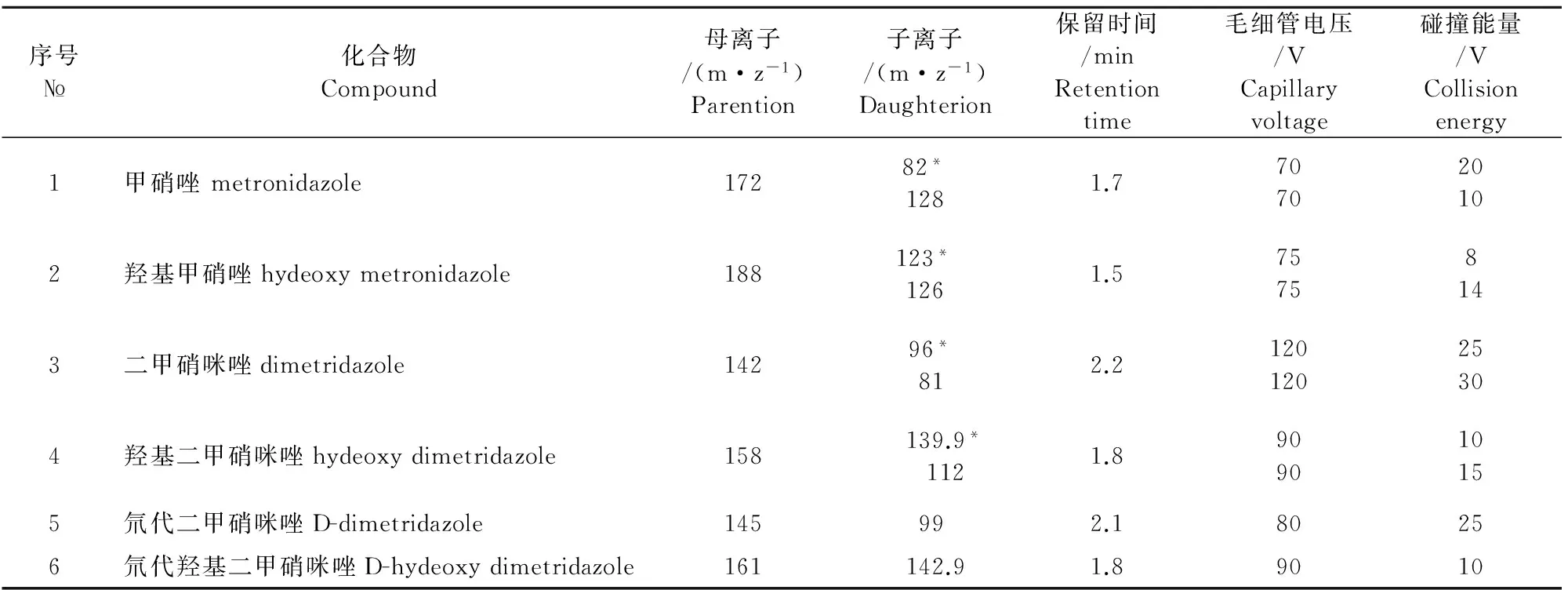
表1 甲硝唑、二甲硝咪唑及代谢物的部分质谱参数
注:*定量离子。
Note: *quantitative ion.
2 结果
2.1 色谱、质谱条件
分别配制500 μg/L的甲硝唑和二甲硝咪唑及代谢物标准品,选用正离子模式扫描,优化各化合物的毛细管电压和碰撞能量,以响应值最高的两个碎片离子和母离子组成定量离子对和定性离子对,并进行MRM参数的优化,4种目标物的部分质谱参数见表1,总离子流图见图1。
2.2 线性范围、检出限与定量下限
对4种目标分析物质量浓度在0.20 μg/L~50 μg/L之间的系列混合标准溶液进行测定,甲硝唑、羟基甲硝唑及羟基二甲硝咪唑以氘代羟基二甲硝咪唑为内标,二甲硝咪唑以氘代二甲硝咪唑为内标,各化合物定量离子与内标校正离子峰面积之比的平均值(y)对其质量浓度 (x,μg/L)绘制标准曲线,其相关系数(r)均大于0.995。在血清和尿液中添加系列浓度的混合标准物质,以信噪比(S/N)为3作为方法的检出限(LOD),以信噪比(S/N)为10作为方法的定量下限(LOQ),甲硝唑和二甲硝咪唑及代谢物的LOD为0.20 μg/L,LOQ为0.50 μg/L。4种目标分析物的回归方程、相关系数、线性范围、检出限及定量下限见表2。
2.3 回收率与精密度
在空白样品中添加甲硝唑和二甲硝咪唑及代谢物定量下限1、2、10倍3个水平的混合标准物质进行加标回收试验。以回收率结果表示方法准确度,回收率的相对标准偏差(RSD)表示方法的精密度,每个水平平行测定6个样品,测定结果见表3。结果显示,甲硝唑和二甲硝咪唑及代谢物在3个加标水平下的平均回收率分别为89.3%~108.2%,相对标准偏差(RSD)分别为2.63%~10.0%。
3 讨论
3.1 样品提取条件的优化
对于血液和尿液中硝基咪唑类药物的提取,适宜的溶剂有有乙酸乙酯[4,12]、二氯甲烷、甲苯及一些混合提取剂。向空白血液和尿液中添加混合标准溶液,使每种待测物浓度为20 ng/mL,分别用乙酸乙酯、二氯甲烷、甲苯进行提取。结果表明,二氯甲烷对硝基咪唑类药物的提取回收率在51.0%~56.4%之间,而乙酸乙酯和甲苯对4种药物的提取率均超过了75%以上,证明乙酸乙酯和甲苯都可以作为提取溶剂,考虑到乙酸乙酯具有更好的环境和安全因素,更有利于分离,最终确定用乙酸乙酯作为提取溶剂。文献报道硝基咪唑类药物在碱性条件下有一定分解,同时发现目标分析物在正离子模式下质谱响应值更好,故本试验设计了乙酸乙酯提取后在不同盐酸比例下对目标分析物提取效率的影响,结果发现,不同的盐酸比例并不能明显的提高甲硝唑和二甲硝咪唑及代谢物的提取效率,所以最终选择只使用乙酸乙酯提取。血清中较多的蛋白质会干扰目标化合物的分析,降低了质谱检测的灵敏度,应该在前处理过程中除去。关于血液中蛋白质的沉淀方法,使用较多的有盐析法,有机溶剂沉淀法,等电点沉淀法和金属离子沉淀法等。但是,等电点沉淀法比较繁琐,血样较少时不利于操作处理;金属离子沉淀法容易污染质谱设备;盐析法有一定的可逆性,而乙腈沉淀蛋白质比较方便,规避了以上缺点,最终选用用乙腈来沉淀蛋白。
3.2 样品净化条件的优化
乙酸乙酯提取液中依然含有一定量极性的杂质,如蛋白质、脂肪等脂溶性物质,尤其是血清样品,基质成分比尿液复杂,为提高方法的灵敏度,本文选用混合型阳离子交换柱净化。考察了提取液过MCX、PCX两种净化柱对目标物回收率和稳定性的影响,发现MCX和PCX净化柱4种目标分析物的平均回收率分别为93.2%和92.1%,因MCX和PCX净化柱回收率差距不明显,从性价比考虑最终选用PCX净化柱。PCX柱属于强阳离子交换柱,在水相偏酸的环境下对目标物有更好的吸附效果,试验进一步考察了甲酸、乙酸及盐酸溶解目标物经PCX柱净化对回收率的影响。提取液经氮气吹干,分别用0.1 mol/L 的甲酸、乙酸及盐酸溶解,经PCX柱净化后经质谱检测,试验发现3种酸性溶液均能将目标物回收率提高到80%以上,但盐酸质谱噪音低,灵敏度比甲酸及乙酸高20%以上,最终选用盐酸溶液溶解。

图1 混合标准溶液(0.5 μg/L)中甲硝唑、二甲硝咪唑及代谢物的多反应监测色谱图
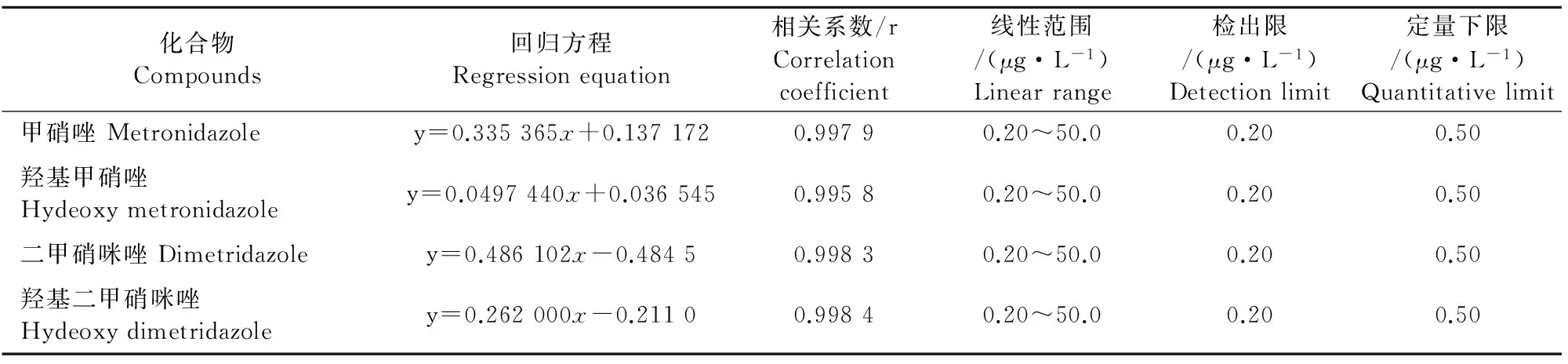
化合物Compounds回归方程Regressionequation相关系数/rCorrelationcoefficient线性范围/(μg·L-1)Linearrange检出限/(μg·L-1)Detectionlimit定量下限/(μg·L-1)Quantitativelimit甲硝唑Metronidazoley=0.335365x+0.1371720.99790.20~50.00.200.50羟基甲硝唑Hydeoxymetronidazoley=0.0497440x+0.0365450.99580.20~50.00.200.50二甲硝咪唑Dimetridazoley=0.486102x-0.48450.99830.20~50.00.200.50羟基二甲硝咪唑Hydeoxydimetridazoley=0.262000x-0.21100.99840.20~50.00.200.50
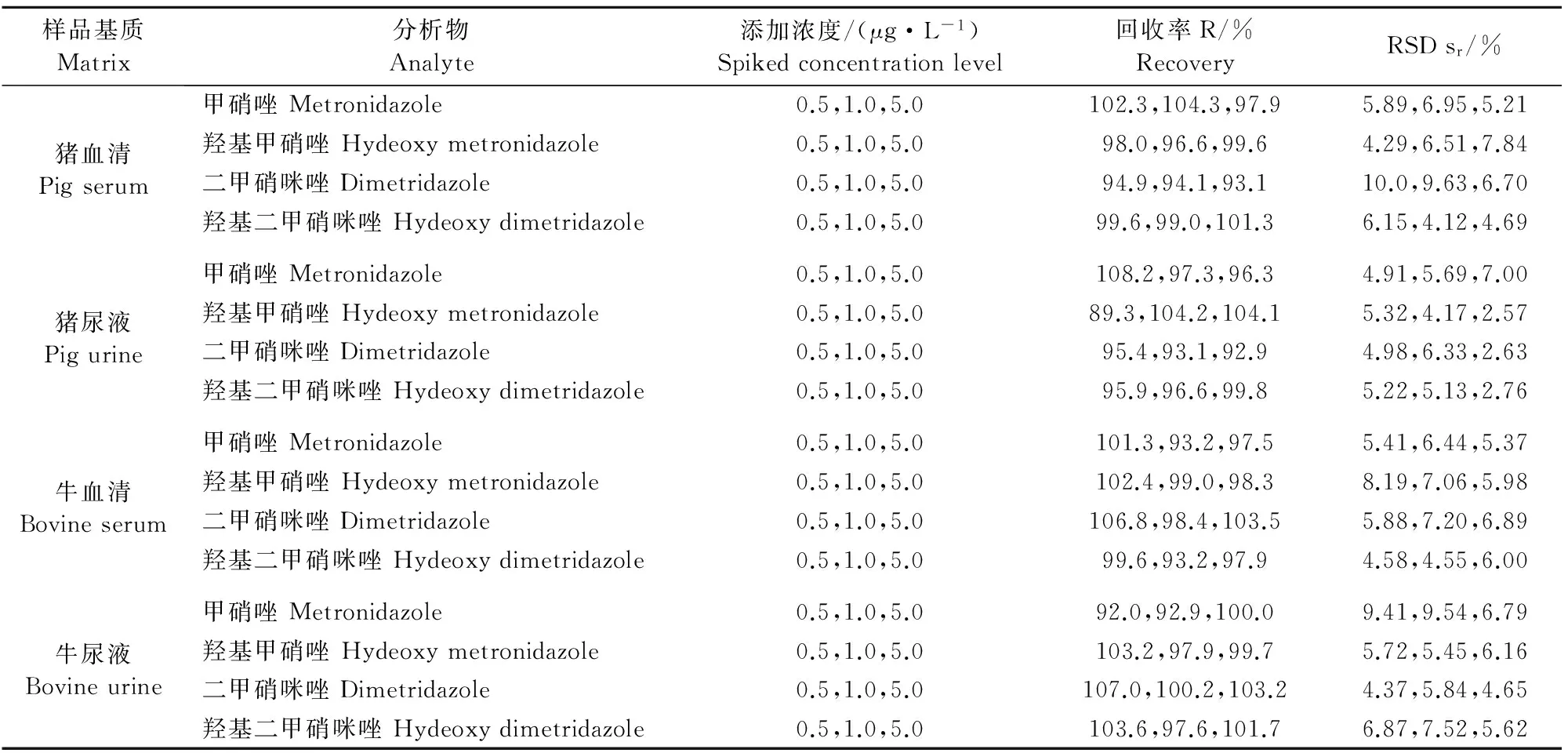
表3 检测方法的添加精密度和平均回收率(n=6)
3.3 色谱分离条件的优化
硝基咪唑类药物及其代谢物是一类具有5-硝基取代咪唑杂环结构的化合物,质量数偏小,血清和尿液基质复杂,色谱分离过程中干扰物较多,因此色谱优化的关键在于提高目标分析物和类似物的分离度。比较了甲醇和乙腈作为流动相对甲硝唑和二甲硝咪唑及代谢物的影响,发现使用乙腈为流动相4种目标物的MRM的提取离子流图峰型尖锐对称,灵敏度更高,保留时间稳定。硝基咪唑类药物属于碱性离子,甲酸的加入有助于促进质子离子化,提高分析灵敏度,试验对比了水相中加入1、2、4 mL/L 3种比例的甲酸溶液,质谱发现1 mL/L甲酸的加入可以提高目标物的响应值,再增加甲酸比例对目标物响应值影响不明显。
由于硝基咪唑类药物具有性质稳定、价格低,既能杀灭及预防厌氧菌和病原虫,也可以促进禽畜类的生长。因此,依然被一些违规养殖场作为抗菌药物备选使用。然而药物的使用会造成其在动物组织中聚集,从而直接或间接地通过食物链的作用对人体产生毒副作用,增加致突变和致癌性风险[11-12]。因此,硝基咪唑类药物的残留及其带来的食品安全问题已引起消费者的广泛关注。目前,在国内关于硝基咪唑类药物多残留检测方法的报道有很多,徐娟等[8]建立了采用超高效液相色谱-串联质谱同时检测口腔卫生用品(牙膏及漱口水)中甲硝唑、替硝唑、奥硝唑、二甲硝咪唑及洛硝唑的方法,该方法可靠、稳定,可满足口腔卫生用品中硝基咪唑类药物含量检测与确证的需要。张鸿伟等[9]报道了采用液相色谱-四极杆/离子阱串联质谱建立了蜂蜜中痕量硝基咪唑类药物( 甲硝哒唑、咯硝哒唑、二甲硝咪唑、异丙硝唑) 及其羟基代谢物( 2-羟甲基-1-甲基-5-硝基咪唑、羟基甲硝唑及羟基异丙硝唑) 残留的快速测定方法。方法快速简便、灵敏度高、重现性好、适用性强,检测限量满足国内外相关法规要求,可有效用于实验室日常分析和残留监控检测。郭菁等[14]建立了高效液相色谱-电喷雾串联质谱快速测定水产品中硝基咪唑类兽药甲硝哒唑、洛硝哒唑、迪美硝唑及其代谢物羟基甲硝唑、羟基二甲硝咪唑残留量的分析方法。该方法操作快速、高效、定量重复性好,适用于水产品中硝基咪唑类药物残留的检测。孟茜等[15]建立了化妆品中16种抗真菌类化合物(克霉唑、联苯苄唑、特比萘酚、酮康唑、伊曲康唑)及硝基咪唑类化合物(甲硝唑、异丙硝唑、苯酰甲硝唑、二甲硝咪唑、替硝唑、奥硝唑、特尼哒唑、洛硝哒唑、氯甲硝咪唑、4-硝基咪唑及塞克硝唑)的高效液相色谱-串联质谱同时检测的方法。该方法快速简便,适用于化妆品中抗真菌类和硝基咪唑类等16种抗生素含量的测定。目前尚未发现同时测定血清和尿液中甲硝唑和羟基甲硝唑及两者代谢物残留量的报道,本文建立了血清和尿液中甲硝唑、羟基甲硝唑、二甲硝咪唑及羟基二甲硝咪唑的高效液相色谱-串联质谱联用的检测方法。该方法具有简便、快速、灵敏、准确、实用性强等优点,可以随时对企业饲养的食用动物进行监控,降低检测成本,为政府监管部门提供技术支撑。
[1] 李小桥,武 煊,李玉平,等.牛肉组织中硝基咪唑类药物残留检测方法研究[J].安徽农学通报,2013,19(9):28-30.
[2] Mahugo-Santana C,Sosa-Ferrera Z,Torres-Padrón M E,et al.Analytical methodologies for the determination of nitroimidazole residues in biological and environmental liquid samples:A review[J].Anal Chim Acta,2010,665(2):113-122.
[3] EEC2377/90关于确定动物源性食品中兽药制品最高残留限量程序的规定.国家质量监督检验检疫总局.欧盟食品卫生法规汇编[M].山东青岛:中国海洋大学出版社,2003:819.
[4] 中华人民共和国农业部公告第193号.农业部兽医局.2002.http://www.moa.gov.cn/zwllm/tzgg/gg/201104/t20110422-1976324.htm.
[5] 傅 哲,彭 智,肖志勇.气相色谱法测定动物血中甲硝唑浓度及药动学研究[J].长沙医学院学报,2010(12):5-9.
[6] 冯学忠,黎华鹏,盘苑芷,等.HPLC同时测定复方二甲硝咪唑可溶性粉中地美硝唑、甲氧苄啶、磺胺对甲氧嘧啶的含量[J].动物医学进展,2011,32(7):55-60.
[7] 秦 松,张 蓓.RP-HPLC法测定人工牛黄甲硝唑胶囊中甲硝唑的溶出度[J].中国医药科学,2015,5(21):48-53.
[8] 徐 娟,陈 捷,邵琳智,等.超高效液相色谱-串联质谱法同时检测口腔卫生用品中的硝基咪唑类药物[J].色谱,2011,29(5):450-453.
[9] 张鸿伟,简慧敏,林黎明,等.液相色谱-四极杆/离子阱质谱快速测定蜂蜜中痕量硝基咪唑类药物及其代谢物残留[J].分析测试学报,2012,31(7):763-770.
[10] 张 怡,李艳芹,赖丽虹,等.液相色谱-串联质谱法测定猪血浆和尿液中8种同化激素的残留[J].动物医学进展,2015,36(6):86-90.
[11] Hanafi A,Lee W C,Loke M F,et al.Molecular and proteomic analysis of levofloxacin and metronidazole resistantHelicobacterpylori[J].Front Microbiol,2016(7):1-12.
[12] Jarrad A M,Debnath A,Miyamoto Y,et al.Nitroimidazole carboxamides as antiparasitic agents targetingGiardialamblia,Entamoebahistolyticaand,Trichomonasvaginalis[J].Eur J Med Chem,2016,14(120):353-362.
[13] Lee J S,Cho S H,Lim C M,et al.A Liquid chromatography-tandem mass spectrometry approach for the identification of mebendazole residue in pork,chicken,and horse[J].PLoS One,2017,12(1):1-13.
[14] 郭 菁, 丁立平, 吴文凡, 等. 高效液相色谱-电喷雾串联质谱法同时测定水产品中硝基咪唑类化合物及其代谢物残留[J]. 分析测试学报, 2015, 34(1):28-34.
[15] 孟 茜, 郑 荣, 王 柯. 高效液相色谱-串联质谱法测定化妆品中16种抗真菌类和硝基咪唑类化合物[J]. 中国卫生检验杂志, 2016, 26(6):784-788.
Residue Determination of Nitroimidazoles in Sera and Urine by High Performance Liquid Chromatography-tandem Mass Spectrometry
ZHANG Jin-ling1,GUO Li-qiang1,SUN Jun1,WU Cui-ling2
(1.WeifangEntry-ExitInspectionandQuarantineBureau,Weifang,Shandong,261041,China; 2.AgilentTechnologiesCo.LTDinChina,Beijing,100102,China)
A method for the determination of metronidazole,hydroxy metronidazole,dimetridazole and hydroxy dimetridazole in sera and urine by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) was established.The target compounds were purified with solid phase extraction column of PCX after extraction by ethyl acetate.Then the samples was separated by an Eclipse Plus C18column(3.0 mm×100 mm,1.8 μm)and analyzed by HPLC-MS/MS.The calibration curves were linear in the range of 0.2 μg/L-50 μg/L for metronidazole,hydeoxy metronidazole,dimetridazole and hydeoxy dimetridazole with correlation coefficient (r) more than 0.995.The LOD was 0.2 μg/L and the LOQ was 0.5 μg/L.The average recoveries (n=6) in feed samples at three spiked levels(1 times,2 times,10 times) ranged from 89.3%-108.2% with relative standard deviations (RSDs) of 2.63%-10.0%.The method was sensitive,convenient and accurate,and could satisfy the demand of detection and analysis of nitroimidazoles in serum and urine samples.
high performance liquid chromatography-tandem mass spectrometry; serum; urine; residue analysis; nitromidazole
2016-11-16
山东出入境检验检疫局科研基金项目(SK201349);潍坊市2015年科学技术发展计划项目(2015ZJ1101)
张金玲(1973-),女,山东高密人,高级兽医师,兽医硕士,主要从事食品分析与检测研究。
S859.7
A
1007-5038(2017)07-0056-06
猜你喜欢
农产品加工(2022年14期)2022-11-17
中学生学习报(2021年20期)2021-11-24
鞍钢技术(2021年3期)2021-06-11
化工管理(2021年7期)2021-05-13
中国农资(2016年1期)2016-12-01
西安工程大学学报(2016年3期)2016-06-05
中国卫生标准管理(2015年2期)2016-01-14
西南军医(2015年2期)2015-01-22
食品安全导刊(2014年8期)2014-10-21
中国药业(2014年12期)2014-06-06
