
黄芩苷对大鼠口服非索非那定的药动学影响
2017-08-09 01:28王斌杨锐李思聪张敏李金良李旭廷
中国兽药杂志 2017年7期
王斌,杨锐 ,李思聪,张敏,李金良,李旭廷*
(1.四川省畜牧科学研究院兽药研究所,成都 610066;2.动物遗传育种四川省重点实验室,成都 610066)
黄芩苷对大鼠口服非索非那定的药动学影响
王斌1,2,杨锐1,2,李思聪1,2,张敏1,2,李金良1,2,李旭廷1,2*
(1.四川省畜牧科学研究院兽药研究所,成都 610066;2.动物遗传育种四川省重点实验室,成都 610066)
为了研究黄芩苷对大鼠口服非索非那定药动学及P-糖蛋白(P-gp)表达的影响,将SD大鼠36只随机分为A、B、C三组,连续灌胃7 d,A组给予黄芩苷200 mg/kg,B组蒸馏水空白对照,C组维拉帕米药物对照(10 mg/kg),第8天灌胃后2 h,各组随机取6只大鼠处死,解剖并迅速取出肝脏、空肠,荧光定量PCR测定各组织P-gp的mRNA表达水平。各组另外6只大鼠灌胃后同时给予非索非那定(30 mg/kg),按时间点连续采集血样,采用高效液相色谱法测定非索非那定血药浓度。结果表明,黄芩苷对非索非那定的吸收有明显促进作用,主要表现在黄芩苷组非索非那定峰浓度(Cmax)比空白对照组显著增加17.04%(P<0.05),曲线下面积AUC(0-12)显著增加19.23%(P<0.05),黄芩苷减少了大鼠空肠和肝脏P-gp的表达(P<0.05)。结论:黄芩苷能下调大鼠P-gp表达,增加P-gp底物非索非那定在大鼠体内的生物利用度,是一种P-gp抑制剂。
黄芩苷;非索非那定;药动学;荧光定量;P-糖蛋白
黄芩苷是黄芩中主要活性成分,属于黄酮类化合物,具有抗病毒、抗炎、免疫调节等作用[1-2]。黄芩苷与多种药物存在相互作用,其与化学药的相互作用问题越来越受到重视[3-5]。P-糖蛋白(P-glycoprotein,P-gp)是由1280个氨基酸组成的跨膜蛋白,在药物吸收、分布、代谢及排泄方面有重要的作用[6]。含有黄芩及黄芩苷的中药制剂在兽医临床应用非常广泛,研究黄芩苷对药物转运蛋白P-gp的影响对指导临床合理用药具有重要意义。本研究以非索非那定药动学参数作为体内P-gp活性的探针,通过观察黄芩苷对非索非那定大鼠体内药动学及肝脏和空肠P-gp表达的影响来探讨黄芩苷对P-gp活性的影响,为临床黄芩苷的药物配伍使用提供科学依据。
1 材 料
1.1 仪器 UltiMate 3000型高效液相色谱仪,美国戴安公司;WH-3微型漩涡混合仪,上海泸西分析仪器厂;Eppendorf MiniSpin plus离心机,德国Eppendorf 公司;CQ250超声洗涤器,上海超声仪器厂;MD200-1氮吹仪,杭州奥盛仪器有限公司。H6-1微型电泳槽,上海精益有机玻璃制品仪器厂;凝胶成像系统,北京君意;T6紫外分光光度计,北京普析通用仪器有限公司;PCR反应扩增仪,BIO公司; StepOne型荧光定量PCR仪,ABI。
1.2 试剂与药品 非索非那定对照品(批号:100852-201202),非那西丁对照品(批号:100095-201205),维拉帕米对照品(批号:100223-200102),黄芩苷对照品(批号:110842-201207),对照品购自中国食品药品检定研究院,甲醇、乙腈为色谱级,其它试剂为分析纯。Trizol提取试剂盒,4S Red Plus核酸染色剂,第一链cDNA合成试剂盒,上海生工生物工程有限公司。
1.3 试验动物 健康清洁级SD雄性大鼠,体重(200±20) g,由成都达硕实验动物有限公司提供,合格证号:SCXK(川)2013-24。整个试验期间,于室温20 ℃~24 ℃下饲养,每天供给饲料及纯化水,采血前12 h限饲。
2 方 法
2.1 试验分组 体重相近雄性大鼠36 只,随机分为3 组,每组12 只。灌胃给药,A组黄芩苷200 mg/kg;B组灌注相同体积的蒸馏水,C组灌注维拉帕米10 mg/kg,每日1次,所有受试大鼠连续灌胃7 d,12 h禁食后,第8天,各组随机挑选6只大鼠,灌药后2 h断颈处死,解剖并迅速取出肝脏、空肠,样品置液氮冻存待检。各组另外6只灌胃后,立即口服非索非那定30 mg/kg,分别于服药后0、0.083、0.25、0.5、0.75、l、1.5、2、3、4、6、8、12 h断尾采血,3000 r/min离心和分离血浆,-20 ℃保存备用。
2.2 血浆样品的处理 取血浆样品100 μL,加入等体积的乙腈,涡旋3 min,12000转离心5 min,取上清液置于另一离心管中,氮吹仪吹干,用流动相100 μL溶解,涡旋1 min,取20 μL进样。
2.3 液相色谱法测定非索非那定血药浓度 色谱条件:色谱柱:Diamonsil C18(200 mm×4.6 mm,5 μm);流动相:乙腈∶0.5%磷酸二氢钾(pH3.8):三乙胺(30∶70∶0.3);流速:1.0 mL·min-1;UV检测波长:210 nm;柱温:35 ℃;进样量:20 μL。在上述色谱条件下,非索非那定与非那西丁、溶剂峰、血浆及其他组分能完全分开,该方法血浆的定量限为0.054 μg/mL。
血浆样品标准曲线范围在0.054~21 μg/mL之间,线性关系良好。在标准曲线范围内,回收率为99.7%~101.4%,批内变异系数和批间变异系数分别为2.67%~5.64%和3.12~4.85%。
2.4 荧光定量PCR检测大鼠肝、空肠P-gp表达 用于UNIQ-10 柱式 Trizol 总RNA抽提试剂盒提取各组样品RNA,按常规方法进行反转录,反应体系为25 μL,反转录产物-20 ℃保存备用。将cDNA样品稀释8倍作为模板上机检测,最终扩增反应按95 ℃预变性3 min,95 ℃变性7 s,57 ℃退火10 s,72 ℃延伸15 s,共45循环。所有样品检测均含有一个平行样本,并包含1个无模板的阴性对照以排除假阳性结果,以GAPDH 管家基因作为内标,对基因P-gp的mRNA表达进行相对定量分析,将所得Ct按照2-(△△Ct)方法进行计算分析统计结果。

表1 实时荧光定量RT-PCR引物序列Tab 1 Primer sequence of real time fluorescent quantitative RT-PCR
2.5 数据分析 采用药动学分析软件DAS3.0,以非房室模型处理血浆浓度-时间数据,获得药动学参数,数据以平均数±标准差(x±s)表示,组间数据采用SPSS11.0统计软件进行t 检验。
3 结 果
3.1 药物动力学结果 各组大鼠体内的非索非那定血药浓度-时间曲线见图1,主要药物动力学参数结果见表2。结果表明,黄芩苷和维拉帕米诱导后,与对照组相比,非索非那定血药浓度-时间曲线下面积AUC(0-12)显著增加(P<0.05),达峰浓度(Cmax)显著升高(P<0.05),达峰时间(Tmax)、半衰期(t1/2)、平均驻留时间MRT(0-12)、清除率(CLZ/F)各组差异不显著。
3.2 大鼠肝、空肠P-gp蛋白mRNA的表达 荧光定量RT-PCR检测结果见图2,维拉帕米和黄芩苷诱导对大鼠肝、肠的P-gp mRNA表达量有下调作用(P<0.05),且维拉帕米的下调程度大于黄芩苷。
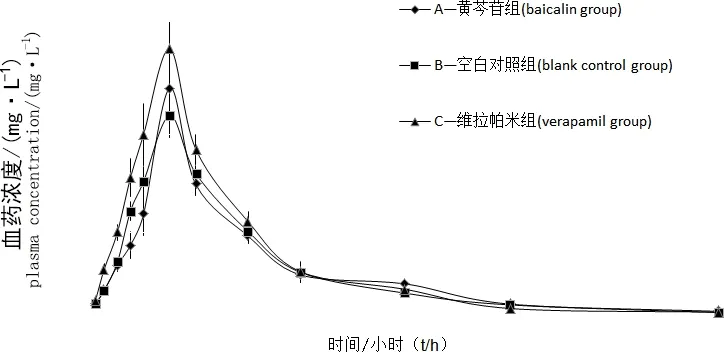
图1 各组灌胃后非索非那定的血药浓度-时间曲线 Fig 1 Plasma concertration-time curves of fexofenadine in the different groups(x±s,n=6)
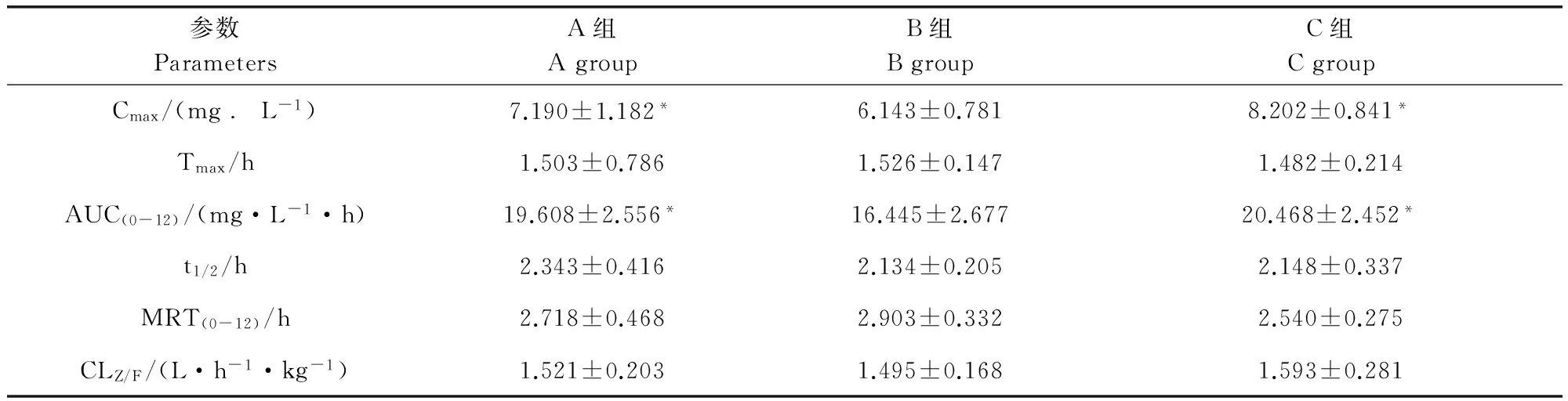
参数ParametersA组AgroupB组BgroupC组CgroupCmax/(mg﹒L-1)7.190±1.182*6.143±0.7818.202±0.841*Tmax/h1.503±0.7861.526±0.1471.482±0.214AUC(0-12)/(mg·L-1·h)19.608±2.556*16.445±2.67720.468±2.452*t1/2/h2.343±0.4162.134±0.2052.148±0.337MRT(0-12)/h2.718±0.4682.903±0.3322.540±0.275CLZ/F/(L·h-1·kg-1)1.521±0.2031.495±0.1681.593±0.281
*0.05水平上差异显著
*Significant difference at 0.05 levels
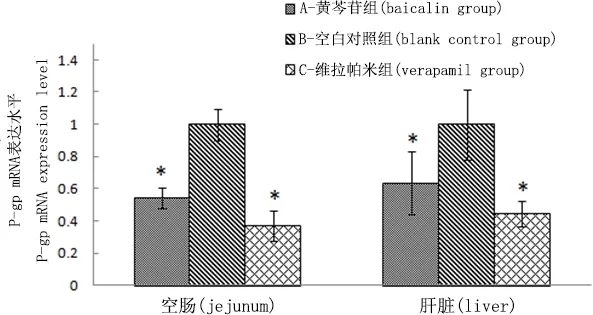
* 0.05水平上差异显著* Significant difference at 0.05 levels图2 各试验组大鼠肝、空肠P-gp mRNA表达水平(x±s,n=6)Fig 2 expression level of P-gp mRNA in the live and jejunum of different group (x±s,n=6)
4 讨 论
黄芩苷为中药黄芩的主要活性成分之一,以黄芩提取物入药的制剂在兽医临床应用广泛,如双黄连注射液、银黄注射液、清开灵注射液等,且多以中西药配伍使用[7],联合使用的药物大多具有药效的协同作用,同时也可能有相似代谢或转运途径,表现为药动学特征的改变,P-gp介导的相互作用最为常见。基于P-gp抑制机制的相互作用往往会使其底物的疗效增加,生物利用度提高,因此,对P-gp抑制剂药物的研究,在指导临床联合用药及解决多药耐药问题都具有重要意义。
Tsuruo等[8]证实维拉帕米对P-gp有抑制作用,可阻碍药物外排,提高细胞内药物浓度,改善治疗药物的体内生物利用度,而非索非那定是药物转运体P-gp 和有机阴离子转运体的底物,由于它在体内不被代谢,常用来作为体内评价P-gp 转运行为的探针药物[9]。本研究以维拉帕米作为阳性对照药物,以非索非那定作为体内P-gp的探针药物,通过观察黄芩苷对非索非那定药动学的影响,探讨黄芩苷对P-gp的影响。结果显示,维拉帕米对非索非那定主要药动学参数影响明显,Cmax和AUC(0-12)较空白组显著增加,表明维拉帕米提高了非索非那定在大鼠体内的生物利用度,呈现出P-gp抑制剂的特性。黄芩苷对非索非那定的药动学参数的改变趋势与维拉帕米一致,提示黄芩苷可能也是P-gp抑制剂。韦灵玉等[10]将黄芩提取液大鼠连续灌胃一周,体外评价P-gp底物罗丹明123和非P-gp底物荧光素钠经大鼠空肠黏膜吸收方向和分泌方向的转运变化,发现黄芩提取液对P-gp活性具有抑制作用。范岚[11]研究了黄芩苷对P-gp底物他林洛尔药代动力学影响,表明黄芩苷能显著提高他林洛尔Cmax和AUC(0-12),显示黄芩苷可以抑制P-gp活性,与本研究结果一致。 除探针药物外,利用荧光定量PCR开展药物转运体、代谢酶的mRNA表达也是研究药物-药物影响和药物代谢的重要方法。Miao Q等[12]利用Caco-2细胞比较了黄芩素和黄芩苷对的P-gp活性和表达水平的影响,发现黄芩素对P-gp活性和表达水平抑制作用明显,而黄芩苷对P-gp的表达无影响。本研究选择大鼠进行体内试验,荧光定量PCR检测诱导给药后大鼠的肝脏和肠道P-gp mRNA表达量。结果显示,黄芩苷连续诱导给药,可下调大鼠肝脏和肠道P-gp的表达,增加药物在肠道吸收,提高药物的生物利用度,这与药动学试验结果一致。大鼠体内试验结果和其他学者体外试验结果的差异,应与大鼠口服黄芩苷后,在肠道内大部分被菌群转化为黄芩素后再吸收有关[13]。
综上所述,黄芩苷通过下调药物转运蛋白P-gp的表达来提高P-gp底物在大鼠体内的吸收,提高了药物的生物利用度,然而大鼠体内存在种类众多的转运蛋白,且药物的吸收代谢还受到肝药酶的影响[14],因此尚需对此进行进一步的研究,更加明确其作用机制,为黄芩苷在临床的应用奠定基础。
[1] 文敏, 李雪, 付守廷, 等. 黄芩苷药理作用研究新进展[J]. 沈阳药科大学学报,2008,25(2):158-162.
Wen M, Li X, Fu S T. New research progress in pharmacological activities of baicalin[J]. Journal of Shenyang Pharmaceutical University,2008,25(2):158-162.
[2] 于宜平, 张艳, 李红, 等. 黄芩苷对角叉菜胶致热大鼠解热作用的PK-PD模型研究[J]. 中草药, 2014,45(4): 527-531.
Yu Y P, Zhang Y, Li H,etal. Pharmacokinetic-pharmacodynamic study on antipyretic effects of baicalin on carrageenan-induced pyrexia of rats[J]. Chinese Traditional and Herbal Drugs, 2014,45(4): 527-531.
[3] Feng J, Xu W, Tao X,etal. Simultaneous determination of baicalin, baicalein, wogonin, berberine, palmatine and jatrorrhizine in rat plasma by liquid chromatographytandem mass spectrometry and application in pharmacokinetic studies after oral administration of traditional Chinese medicinal preparations containing scutellaria-coptis herb couple[J]. J Pharm Biomed Anal, 2010, 53(3): 591-598.
[4] Fong S Y K, Wong Y C, Zuo Z. Development of a SPE-LC/MS/MS method for simultaneous quantification of baicalein, wogonin, oroxylin A and their glucuronides baicalin, wogonoside and oroxyloside in rats and its application to brain uptake and plasma pharmacokinetic studies[J]. J Pharma Biomed Anal, 2014, 97: 9-23.
[5] Zhou S F, Zhou Z W, Li C G,etal. Identification of drugs that interact with herbs in drug development[J]. Drug Discov Today 2007:12:664-673.
[6] 蒋学华. 临床药动学[M]. 北京: 高等教育出版社,2007.60.
Jiang X H. Clinical Phamacokinetics[M]. Beijing: Higher Education Press, 2007.60.
[7] 张魁华, 陈瑞爱, 方炳虎, 等. 中兽药注射剂与其他兽用注射剂的配伍[J]. 中国兽药杂志,2007,41(10):50-52.
Zhang K H, Chen R A, Fang B H,etal. Studies on the Compatibility of Tradtional Chinese Veterinary Medical Injection and Other Veterinary Medical Injection[J]. Chinese Journal of Veterinary Drug, 2007,41(10):50-52.
[8] Tsuruo T, lida H, Tsukagoshi S,etal. Increased accumulation of vincristine and adriamycin in drug-resistant P388 tumor cells following incubation with calcium antagonists and calmodulin inhibitors[J]. Cancer Research, 1982,42(11): 4730-4733.
[9] Russell T, Stohz M, Weir S. Pharmacokinetics,pharmacodynamics and tolerance of single-and multiple-dose fexofenadine hydrochloride in healthy male volunteers[J].Clin Pharm Ther,1998,64( 6): 612-621.
[10]韦灵玉, 李静, 刘晓亚, 等. 黄连黄芩配伍对大鼠肠黏膜P-gp活性的影响[J]. 中国实验方剂学杂志, 2012,18(08):141-146.
Wei L Y, Li J, Liu X Y,etal. Induction of P-glycoprotein in Rat Jejunum by Combined Use of Coptis-scute Herb[J]. Chinese Journal of Experimental Traditional Medical Formulae, 2012,18(08):141-146.
[11]范岚. 黄芩苷对CYP450代谢酶和药物转运体影响的研究[D]. 湖南:中南大学,2008.
Fan L. Study on Effects of Baicalin on CYP450 Metabolism Enzymes and Drug Transporters[D].Hu Nan: Central South University,2008.
[12]Miao Q, Wang Z, Zhang Y,etal. In vitro potential modulation of baicalin and baicalein on P-glycoprotein activity and expression in Caco-2 cells and rat gut sacs[J]. Pharm Biol, 2016, 54(9):1548-1556.
[13]刘太明, 蒋学华, 张梅娟, 等. 黄芩苷和黄芩素的大鼠在体肠吸收特性[J].中国药学杂志, 2016, 41(23):1784-1787.
Liu T M, Jiang X H, Zhang M J,etal. Absorption Characteristics of Baicalin and Baicalein in Rat Small Intestine[J]. Chin Pharm J, 2006 December, 41(23):1784-1787.
[14]Eloranta J J, Kullak-Ublick G A. Coordinate transcriptional regulation of bile acid homeostasis and drug metabolism[J]. Arch Biochem Biophys,2005, 433(2): 397.
(编辑:陈希)
Effects of Baicalin on Pharmacokinetics of Fexofenadine in Ratsinvivo
WANG Bin1,2, YANG Rui1,2,LI Si-cong1,2, ZHANG Min1,2,LI Jin-liang1,2, LI Xu-ting1,2*
(1.InstituteofVeterinaryPharmacology,SichuanAnimalScienceAcademy,Chengdu610066,China; 2.SichuanAnimalBreedingandGeneticsKeyLaboratoryofSichuanProvince,Chengdu610066,China)
LIXu-ting,E-mail: 676049640@qq.com
To investigate the effects of baicalin on the pharmacokinetics of fexofenadine and the expression level of P-gp in rats,36 SD rats were randomly divided into A,B and C groups, rats in A group were oral administered with baicalin(200 mg/kg), B with distilled water and C with verapamil (10 mg/kg) respectively for 7 d. On the 8th d, After 2 h of administering, 6 rats in each group were killed .The liver and jejunum were collected respectively to evaluate the P-gp mRNA expression level by real-time fluorescent quantitative PCR. The other rats were administered with fexofenadine(30 mg/kg). The concentrations of fexofenadine in plasma after administration were determined by HPLC. After administered with baicalin, Cmaxand AUC(0-12)of nifedipine were significantly increased, and the level of P-gp mRNA expression in liver and jejunum were decreased. It is concluded that baicalin is an inhibitor of P-gp, which can enhance the bioavailability of nifedipine and decrease the level of P-gp mRNA expression significantly
baicalin;fexofenadine;pharmacokinetics;real time PCR;P-gp
四川省公益性科研院所基本科研业务费项目(SASA2015A16);四川省财政运行专项(SASA2014CZYX010) 作者简介: 王斌,硕士,从事新兽药的开发与应用工作。
李旭廷。 E-mail: 676049640@qq.com
10.11751/ISSN.1002-1280.2017.7.10
2017-02-23
A
1002-1280 (2017) 07-0052-05
S859.79
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
新乡医学院学报(2022年6期)2022-11-23
现代消化及介入诊疗(2022年4期)2022-07-12
世界科学技术-中医药现代化(2021年9期)2021-12-31
医学前沿(2021年18期)2021-04-14
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
中成药(2019年12期)2020-01-04
中成药(2019年12期)2020-01-04
南方周末(2014-09-25)2014-09-25
