
Using induced pluripotent stem cells derived neurons to model brain diseases
2017-08-07 12:56CindyMcKinney
中国神经再生研究(英文版) 2017年7期
Cindy E. McKinney
iPSC Lab/Edward Via College of Osteopathic Medicine ande Gibbs Research Institute, Spartanburg, SC, USA
Using induced pluripotent stem cells derived neurons to model brain diseases
Cindy E. McKinney*
iPSC Lab/Edward Via College of Osteopathic Medicine ande Gibbs Research Institute, Spartanburg, SC, USA
How to cite this article:McKinney CE (2017) Using induced pluripotent stem cells derived neurons to model brain diseases. Neural Regen Res 12(7):1062-1067.
The ability to use induced pluripotent stem cells (iPSC) to model brain diseases is a powerful tool for unraveling mechanistic alterations in these disorders. Rodent models of brain diseases have spurred understanding of pathology but the concern arises that they may not recapitulate the full spectrum of neuron disruptions associated with human neuropathology. iPSC derived neurons, or other neural cell types, provide the ability to access pathology in cells derived directly from a patient’s blood sample or skin biopsy where availability of brain tissue is limiting.us, utilization of iPSC to study brain diseases provides an unlimited resource for disease modelling but may also be used for drug screening for e ff ective therapies and may potentially be used to regenerate aged or damaged cells in the future. Many brain diseases across the spectrum of neurodevelopment, neurodegenerative and neuropsychiatric are being approached by iPSC models.e goal of an iPSC based disease model is to identify a cellular phenotype that discriminates the disease-bearing cells from the control cells. In this mini-review, the importance of iPSC cell models validated for pluripotency, germline competency and function assessments is discussed. Selected examples for the variety of brain diseases that are being approached by iPSC technology to discover or establish the molecular basis of the neuropathology are discussed.
induced pluripotent stem cells; neuron cell models; brain diseases; molecular mechanisms; therapeutics; translational medicine
Accepted: 2017-06-26
Introduction
Yamanako (2008) showed that differentiated somatic cells could be reprogrammed to induced pluripotent stem cells (iPSC). Most commonly, fibroblasts or peripheral blood mononuclear cells (PBMC) are captured from patients with neurodegenerative diseases and used to make iPSC derived neural cells to study disease pathology.e reprogramming methodology requires four transcription factors (KLF4, c-MYC, OCT4 and SOX2) that reset the embryonic state (for details see Yamanaka, 2008). iPSC clones can then be transformed to selected di ff erentiated cell types by adding the necessary growth/differentiation proteins and co-factors to the cell culture medium. iPSCs are particularly useful for studying human neurons and other brain cells that may only be available from autopsy material. Using patient derived iPSC allows the researcher to create de fi ned neuron populations that can be scaled to meet research needs. Neurodegenerative and neurodevelopmental disorders are particularly approachable using iPSC technology as di ff erentiated cell types provide the patient’s genomic context to investigate the etiology of disease. iPSC derived cell models also bridge the gap when no appropriate animal model is available and they o ff er the capability to model neuron subtypes like glia and astrocytes. Consequently, cellular and molecular phenotypes driving neuropsychiatric, neurodevelopmental and neurodegenerative disorders may require a human neuronal cellular model that is able to recapitulate the genetic causes of the pathology and to produce the target cell type for study (Acab and Muotri, 2015). In this short review, the approach and some criteria for obtaining fully characterized human iPSC and neurons to investigate pathology and molecular disruptions of function in neurodegenerative or neurodevelopmental diseases is outlined.
Characterizing Reprogrammed Human iPSC
Protocols that detail the steps necessary to obtain iPSC are available from many sources (Bohl et al., 2016). With rigorous culture practices, iPSCs are readily produced.e iPSC must be fully characterized to assure quality before transforming them to di ff erentiated neurons or other brain cells. Characterization includes: immunocytochemistry for known pluripotency markers, con fi rmation that the iPSCs can differentiate to the three germ layers (endoderm, mesoderm and ectoderm), and gene expression profiles for known pluripotency markers. Common pluripotency markers are SOX2, OCT4, TRA1-60, NANOG and TRA1-81. Antibodies are readily available from several sources to conduct these assays. Figure 1 shows a representative panel of pluripotency results from a Gaucher disease type 2 iPSC clone veri fi cation. High quality iPSC clones will report several pluripotency markers over the surface of the iPSC clone.e morphology of the iPSC clone should have clearly defined margins and no miscellaneous di ff erentiated cells growing from the colony. Multiple, robust medium size colonies grow spread over the culture plate surface.e iPSC within the clone contain very little cytoplasm. A second round of veri fi cation includesthe determination of the iPSC clone’s ability to differentiate into all three germ layers, endoderm, mesoderm and ectoderm. This can be done in two ways: 1) injecting iPSC cells into a mouse and waiting for a tumor (teratoma) to develop (Nelakanti et al., 2015) and then histologically analyzing the tissue types within the tumor or 2) differentiating the cells into the germ layer types using de fi ned mediums containing the necessary di ff erentiation factors.ese bioassays con fi rm the pluripotency of the generated iPSC clones before using them to model disease.
Di ff erentiating hiPSC to Neurons and Assessing Functionality
iPSC may be transduced to form most neural cell types (Begum et al., 2015),e.g., cortical neurons, dopaminergic neurons, astrocytes or oligodendrocytes. This is done by supplying iPSC cultures with the appropriate di ff erentiation and growth factors in culture medium and then maintenance growth in an appropriate de fi ned neural maintenance medium, like Brain Phys (Bardy et al., 2015) while experimental evaluations are ongoing. Initial evaluations include analysis of biomarkers of neuronal differentiation (Nestin, NeuN, and others) by ICC or Western blot. Functional screening of neurons should include their ability to generate electrophysiological properties (see Figure 2B and C). All iPSC lines or iPSC derived neural lines should be authenticated to be free of human infectious agents and mycoplasma. They should also be karyotyped to show that no major chromosome rearrangements have occurred during the reprogramming or di ff erentiation process. With these characterizations and authentication of the cell model accepted, it is appropriate to move to analyzing the speci fi c disease pathology.
Studies Analyzing Neuropathology
iPSC and Inborn Errors of Neuron Metabolism
Lysosomal storage disorders
Neuronal ceroid lipofuscinoses (NCL)
NCL are inherited as recessive lysosomal storage diseases. An estimated 14 di ff erent genes (NCL1-NCL14) are involved where di ff erent mutations present in several forms of NCL in pediatric patients. A Finnish research team has focused on NCL5 (variant Jansky-Bielschowsky disease).e NCL5 protein is a lysosomal glycoprotein of, as yet, unknown function (Uusi-Rauva et al., 2017). It is suggested that NCL5 protein is involved in cholesterol and sphingolipid metabolism as well as endosomal sorting. A NCL5 mouse KO model is available (Kopra et al., 2004; Schmiedt et al., 2012) but it is uncertain if this model completely de fi nes the human pathology since the mice do not replicate the characteristic seizures seen in NLC5 patients. To investigate the nature of the disease and to compare human neurons to animal model findings, iPSCs were produced from a NCL5 patient’s fibroblasts with the Finnish variant mutation (c.1175_1176delAT, p.Y329X) and then di ff erentiated to neurons for pathological evaluation. iPSC derived neurons contained an accumulation of auto fl uorescent material similar to fi ndings in NCL5 patient fibroblasts. NCL5 iPSC derived neurons also sequestered the fluorescent synthetic glycosphingolipid, BODIPY-lactosylceramide, as previously shown in the animal model.is fi nding indicates a disruption in sphingolipid transport from the endo-lysosomes to the Golgi in these neurons (Uusi-Rauva et al., 2017).e NCL5 neurons also show ER stress, potentially due to retention of the mutant protein in the ER. However, the observed ER stress may be a result of a non-lysosomal function for the NCL5 protein.ese studies of disease-relevant NCL5 neurons begin to unravel the role of NCL5 protein function and disease pathology in a previously unavailable human cell model.
Batten disease
Batten disease, another neural ceroid lipofuscinosis (NCL3), results in premature death due to progressive motor and cognitive decline, retinal pigment degeneration and seizures (Lojewski et al., 2014). Abnormal pathology was replicated in multiple non-related CNL3 neural lines generated from patient iPSC. Functional de fi cits are observed for mitochondria, Golgi and the endoplasmic reticulum as well as lysosomal storage in the CNS.ese critical defects were detected prior to the appearance of lysosomal storage, identifying a key cellular dysfunction (Lojewski et al., 2014). The authors point to these iPSC models as a way to screen novel drug therapies and they show proof-of-concept studies that evaluate the e ffi cacy ofthe lipid lowering drugs, fenofibrate and gemfibrozil, on this NCL3 model.
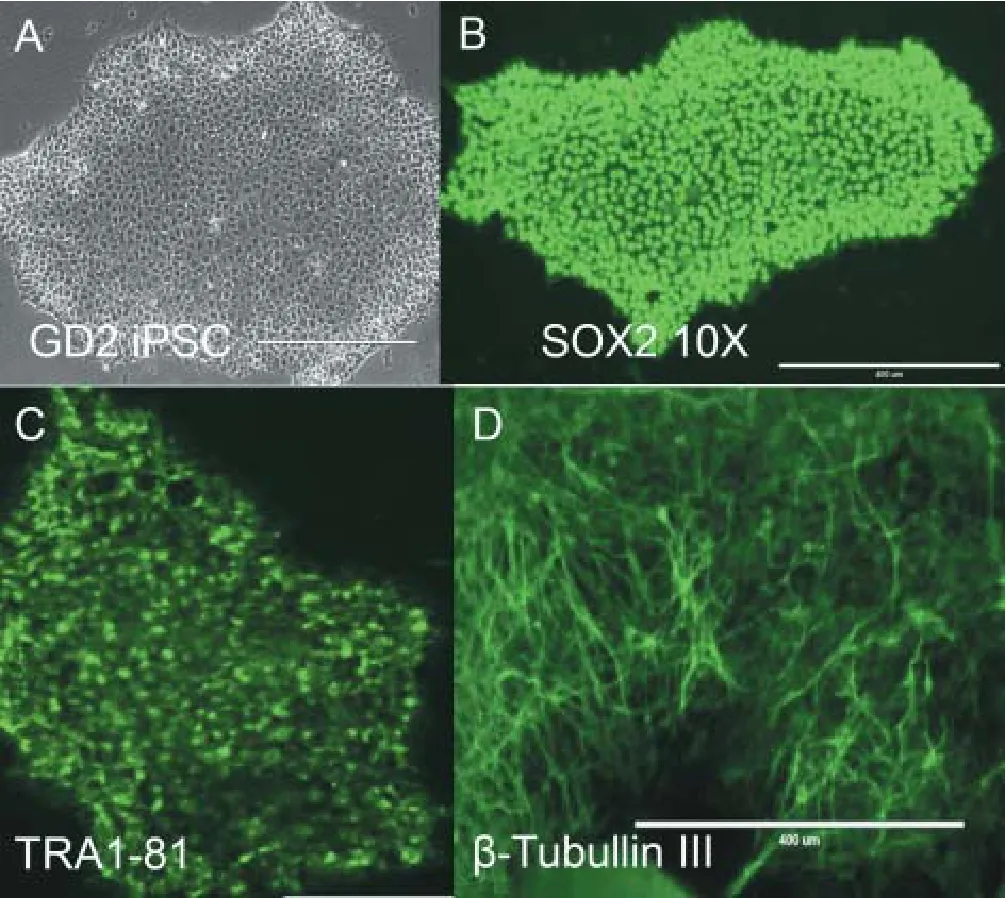
Figure 1 Characterizing induced pluripotent stem cells (iPSC) clones reprogrammed from patient fi broblasts.
Pompe disease
Pompe disease, another neural metabolic disease, is an acid alpha-glucosidase (GAA) deficiency. A neural model was generated from patient iPSC (Higuchi et al., 2014). GAA is the only enzyme that hydrolyzes glycogen to glucose in the acidic environment of the lysosome.us, GAA de fi ciency results in glycogen sequestering and subsequent enlargement of the lysosomes (Lim et al., 2015). Ultrastructure analysis of iPSC derived Pompe neurons recapitulated storage of glycogen granules in the cytoplasm. In a potential enzyme replacement approach, recombinant GAA treatment to correct iPSC derived Pompe neurons showed reduction of storagethat potentially opens a translational approach for therapy. Another lysosomal storage disease model reports iPSC derivation of Niemann-Pick type C1 neurons for study (Trilck et al., 2013).
iPSC and Neurodegenerative Disorders
Parkinson’s disease
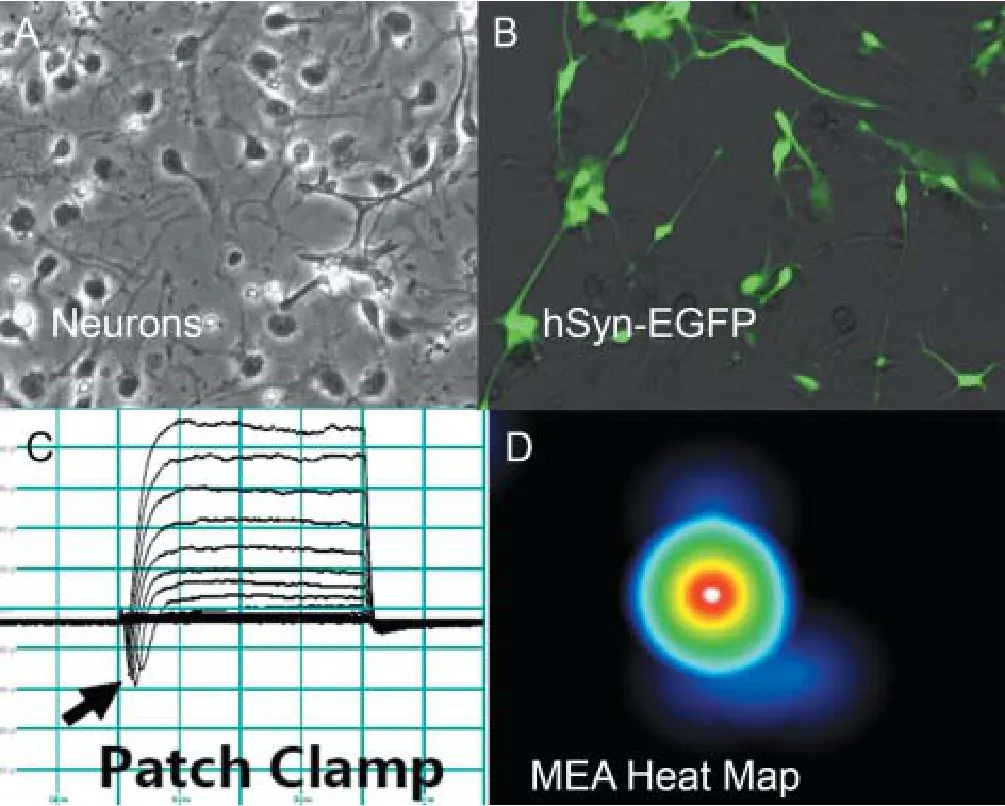
Figure 2 Characterization of Gaucher type 2 (GD2) neurons by biomarkers and functional analysis.
Parkinson’s disease (PD) is a relatively common multifactorial neurodegenerative disease (Sanchez-Danes et al., 2012) characterized by deficits of motor skills. Movement loss is mapped to dysfunction of the dopaminergic neurons (DAn) in the substantia nigra.e loss of DAn is progressive and alpha-synuclein (SCNA) intra-neuronal inclusions, known as Lewy bodies, are seen as the disease progresses.e majority of PD cases are sporadic and approximately 10% of cases are monogenic. The loci involved have documented mutations in the gene for Leucine Rich-Repeat Kinase 2 (LRRK2) and/or SCNA. Early onset PD is associated with mutations in the genes for Parkin, DJ-1, PINK1 and ATP13A2 (Lees et al., 2009). Many current animal models of PD do not demonstrate the key findings of the human disease; thus, PD remains poorly understood.e ability to reprogram patient samples (blood PBMCs or fi broblasts) to iPSC derived DAn provides a means for direct assessment of PD pathology. Several groups have now generated PD neurodegenerative disease models from iPSC that show pathology specific for the human disease (Sanchez-Danes et al., 2012; Fernandez-Santiago et al., 2015). Sanchez-Danes et al. (2012) report differences in neurite outgrowth between control and PD DAn, SCNA accumulation and alterations in autophagic fl ux (LC3-II assay). Fernandez et al. (2015) report that iPSC derived DAn from both sporadic and familial mutations inLRRK2demonstrate large DNA methylation changes in enhancers and transcriptome changes in a regulated gene network. Altered Ca2+dynamics are also reported for PD neurons.
Amyotropic lateral sclerosis (ALS) corrected motor neurons
ALS is a debilitating neurodegenerative adult onset disorder that results in a progressive loss of upper and lower motor neurons while cognitive function remains unchanged. Nocure or clinical treatment halts ALS progression. It is estimated that about 20% of ALS cases are familial and mutations are identi fi ed in more than 20 genes that operate in diverse cellular pathways (Andersen and Al-Chalabi, 2011; Sreedharan and Brown, 2013). Rodent models of ALS (Kobayakawa et al., 2015) are available (SOD1 mouse, G93A), have been studied extensively and have yielded insights into ALS disease pathophysiology. However, as is frequently the case, the relevance of the mouse SOD1 model to human ALS is unclear because of species-speci fi c di ff erences. For example, most rodent models overexpress the mutant ALS related proteins under evaluation at non-physiological levels (Calvo et al., 2012; Cozzolino et al., 2013). Using motor neurons derived from patient iPSC and an isogenic CRISPR corrected iPSC motor neuron line, Bhinge et al. (2017) addressed the question of why motor neurons are targeted in ALS and non-motor neurons are less a ff ected. They report that JUN, a member of the AP1 complex, is expressed at much higher levels in motor neurons. This fi nding led the authors to suggest that JUN has a function in preserving motor neuron homeostasis.In situhybridization data from human spinal cord tissue also shows JUN mRNA at high levels in motor neurons compared to other spinal cord cells and were even higher in spinal tissues from ALS patients (Virgo and de Belleroche, 1995). So, using gene corrected motor neurons derived from iPSC allowed a focused comparison of the diseased and disease corrected cell line to reveal elevated levels of a specific transcription factor (JUN) somehow related to the known SOD1 mutation.
iPSC and Neurodevelopmental Disorders
Autism spectrum disorders (ASD)
ASD are a polygenic set of neurodevelopmental behaviors that share some core symptomology (impairment of social interactions, repetitive behaviors and de fi cits of interpersonal communication) (Acab and Muotri, 2015). About 10–20% of ASD is classi fi ed by known genomic alterations, for example, Rhett Syndrome (Balachandar et al., 2016). Non-syndromic causes of ASD are characterized byde novoand other hereditary mutations.e X-chromosome contains many genes that appear to be involved with intellectual disability that are currently under investigation (Lubs et al., 2012). As with other neurological diseases, the absence of relevant disease models has hindered ASD investigation. Animal models are productive for the study of single gene defects but are currently unable to adequately address complex disease and many do not appropriately address the social and behavior disorders seen in autism. Many monogenic models of ASD are available but iPSC derived neural models from patient samples are especially valuable for non-syndromic ASD (idiopathic) precisely because known genetic mutations are yet to be defined. Griesi-Oliveira et al. (2015) report that iPSC neurons from a protein channel transient receptor potential canonical 6 (TRPC6) mutant contain morphologic and functional alterations when compared with control neurons. TRPC6 may also play an important role in many signaling pathways (e.g., BDNF) and neuronal calcium/calmodulin dynamics (Griesi-Oliveira et al., 2015). Since ASD has a neurodevelopmental etiology, iPSCs can be used to study early human neurogenesis in culture. Investigators compared neurodevelopmental transcriptome profiles from RNASeq of iPSC derived neurons made from both fibroblasts and dental pulp (Chen et al., 2013). Several lncRNAs were seen to become down-regulated during the transition from iPSC to neural progenitor cells while coding genes were up-regulated including many of the HOX genes (DLX1 and POU3F3).ese studies show another example of the utility of iPSC derived neurons to study neurologic disease progression using exomic sequencing to define molecular pathway analysis. These analyses potentially capture alternative splicing defects, up or down regulation of key transcription factors or relevant alterations in miRNA or lncRNA regulators.
IPSC and Neuropsychiatric Diseases
Frontotemporal dementia (FTD)
FTD is a heterogeneous neurodegenerative disorder presenting with cognitive impairment that a ff ects frontal and/ or temporal lobes function of the brain associated with progressive brain atrophy (Rossor et al., 2010). FTD accounts for 6–8% of early onset (< 65 years old) dementia cases. A mutation inCHMP2B(charged microvesicular body protein 2B) found on human chromosome 3 is linked as a cause for frontotemporal dementia type 3 (FTD3) in some familial cases (Zhang et al., 2017).CHMP2Bfunctions as a partner in the endosomal sorting complex required for transport (ESCRT) and when mutated it disrupts endosome-to-lysosome tra ffi cking, and consequently, substrate degradation in the lysosome. Zhang et al. (2017) developed three FTD3 neuronal lines from reprogrammed patient iPSC and gene corrected theCHMP2Bsplice acceptor mutant (31449G > C) to create isogenic control lines. Independent (non-isogenic) controls were also included in the study. FTD3 and isogenic iPSC were differentiated to forebrain specific cortical neurons and further characterized. Electron microscopy showed enlarged endosomes in the FTD3 neurons as previously seen in patient cells. Mitochondria were examined and shown to be cristae-lessand to cluster in the perinuclear region of the neuron. CRISPR corrected FTD3 neurons and independent controls showed the normal distribution of mitochondria in the axons and dendrites as well as the perinuclear region.is fi nding correlated with FTD3 patient’s increased axonal degeneration. Mitochondria in the FTD3 cells also exhibited oxidative stress. RNA-Seq analysis found that the FTD3 transcriptomes were clustered and showed gene expression differences in endosome genes and neurodegeneration genes, such as,LRRK2/Parkinson’s disease andMPOandAPOD/Alzheimer’s disease were down-regulated. In addition, genes involved in iron homeostasis (TRPC6,HFE) showed altered expression pro fi les.us, FTD3 cortical neurons in this study confirmed results from patient tissue including early endosome trafficking defects, mitochondrial stress and suggested that defects in iron homeostasis are part of the FTD3 spectrum.
In another FTD study (Almeida et al., 2013), skin biopsies from patients with the FTD linked repeat expansion GGGGCC in the noncoding region ofC9ORF72, a gene of unknown function, were reprogrammed to iPSC and differentiated to post-mitotic neurons. These FTD neurons showed signi fi cantly elevated p62 levels and increased sensitivity to cellular stress induced by autophagy inhibitors.ese fi ndings showed that key neuropathological features of GGGGCC patients are recapitulated in iPSC-derived human neurons and suggested that compromised autophagy may be a common pathogenic mechanism in neurodegenerative disorders. Other psychiatric disorders that have been approached by iPSC analysis are reviewed in Paşca et al. (2014) where they address the use of iPSC derived neurons to attempt to uncover molecular phenotypes and e ff ective drug treatments.
Conclusion
There is a need for human neuron cell models of disease because of potential species-speci fi c di ff erences with rodent KO and transgenic models. Also, the limited availability of viable neurons from patient brain tissue restricts investigation into the mechanisms of neural pathogenesis. Relevant human models for neurodegenerative, neurodevelopmental, neuropsychiatric disorders and others can be derived from patient somatic cells by reprogramming to iPSC and then deriving neurons. Neural cell models may be generated from the same patient by CRISPR correction of the mutation. Such genetically edited iPSCs are ideal isogenic controls for the patient-derived iPSCs neurons, probing the signi fi cance of the disease-causing mutation in the patient’s own genetic context. iPSC neuron studies provide proof of concept models that allow experimental analysis of disease pathogensis linking them to molecular phenotypes. Human iPSC models provide invaluable accessibility for studying disease progression from early progenitor cells to aged or mature neurons.
Emerging findings in the iPSC derived brain cells suggest, that while mutations are diverse, there may be a shared cellular dysfunction in endolysosomal pathways. Finally, human iPSC derived neurons offer targeted development of drug screening platform(s) and, in the future, they may facilitate the development of new therapies (Griffin et al., 2015; Sugai et al., 2016) across a wide spectrum of neural disorders.ese platforms and potential therapies might be able to treat neurologic disease before clinical symptoms appear and, with a prescreen in iPSC neurons, earlier success in clinical trials might be obtained.
Acknowledgments:
Author contributions:The author contributed figure 1 material from her iPSC lab and panels A and B in fi gure 2 from her lab.
Con fl icts of interest:None declared.
Open access statement:
Contributor agreement:A statement of “Publishing Agreement” has been signed by an authorized author on behalf of all authors prior to publication.
Plagiarism check:This paper has been checked twice with duplication-checking soware ienticate.
Peer review:A double-blind and stringent peer review process has been performed to ensure the integrity, quality and signi fi cance of this paper.
Open peer review report:
Reviewer: Aurel Popa-Wagner, University Medicine Rostock, Germany.
Comments to author: In this review, the author make a survey of available literature with regard human iPSC and iPSC-derived neurons to investigate pathology and molecular disruptions of function in neurodegenerative/neurodevelopmental diseases. While the review is well written, it would be of interest to know if there is any published data on iPSC-derived neurons from patients with co-morbidities.
Acab A, Muotri AR (2015) The use of induced pluripotent stem cell technology to advance autism research and treatment. Neurotherapeutics 12:534-545.
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126:385-399.
Andersen PM, Al-Chalabi A (2011) Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 7:603-615.
Balachandar V, Dhivya V, Gomathi M, Mohanadevi S, Venkatesh B, Geetha B (2016) A review of Rett syndrome (RTT) with induced pluripotent stem cells. Stem Cell Invest 3:52.
Bardy C, van den Hurk M, Eames T, Marchand C, Hernandez RV, Kellogg M, Gorris M, Galet B, Palomares V, Brown J, Bang AG, Mertens J, Bohnke L, Boyer L, Simon S, Gage FH (2015) Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci U S A 112:E2725-2734.
Begum AN, Guoynes C, Cho J, Hao J, Lutfy K, Hong Y (2015) Rapid generation of sub-type, region-speci fi c neurons and neural networks from human pluripotent stem cell-derived neurospheres. Stem Cell Res 15:731-741.
Bhinge A, Namboori SC, Zhang X, VanDongen AMJ, Stanton LW (2017) Genetic correction of SOD1 mutant ipscs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep 8:856-869.
Bohl D, Pochet R, Mitrecic D, Nicaise C (2016) Modelling and treating amyotrophic lateral sclerosis through induced-pluripotent stem cells technology. Curr Stem Cell Reser 11:301-312.
Calvo AC, Manzano R, Atencia-Cibreiro G, Olivan S, Munoz MJ, Zaragoza P, Cordero-Vazquez P, Esteban-Perez J, Garcia-Redondo A, Osta R (2012) Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS One 7:e32632.
Chen J, Lin M, Foxe JJ, Pedrosa E, Hrabovsky A, Carroll R, Zheng D, Lachman HM (2013) Transcriptome comparison of human neurons generated using induced pluripotent stem cells derived from dental pulp and skin fi broblasts. PLoS One 8:e75682.
Cozzolino M, Ferri A, Valle C, Carri MT (2013) Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci 55:44-49.
Fernandez-Santiago R, Carballo-Carbajal I, Castellano G, Torrent R, Richaud Y, Sanchez-Danes A, Vilarrasa-Blasi R, Sanchez-Pla A, Mosquera JL, Soriano J, Lopez-Barneo J, Canals JM, Alberch J, Raya A, Vila M, Consiglio A, Martin-Subero JI, Ezquerra M, Tolosa E (2015) Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med 7:1529-1546.
Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, Nunez Y, Walker MF, Murdoch JD, Sanders SJ, Fernandez TV, Ji W, Lion RP, Vadasz E, Dietrich A, Pradhan D, Song H, Ming GL, Gu X, Haddad G, et al. (2015) Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 20:1350-1365.
Gri ffi n TA, Anderson HC, Wolfe JH (2015) Ex vivo gene therapy using patient iPSC-derived NSCs reverses pathology in the brain of a homologous mouse model. Stem Cell Rep 4:835-846.
Higuchi T, Kawagoe S, Otsu M, Shimada Y, Kobayashi H, Hirayama R, Eto K, Ida H, Ohashi T, Nakauchi H, Eto Y (2014)e generation of induced pluripotent stem cells (iPSCs) from patients with infantile and late-onset types of Pompe disease and the effects of treatment with acid-alpha-glucosidase in Pompe’s iPSCs. Mol Genet Metab 112:44-48.
Kobayakawa Y, Sakumi K, Kajitani K, Kadoya T, Horie H, Kira J, Nakabeppu Y (2015) Galectin-1 de fi ciency improves axonal swelling of motor neurones in SOD1(G93A) transgenic mice. Neuropathol Appl Neurobiol 41:227-244.
Kopra O, Vesa J, von Schantz C, Manninen T, Minye H, Fabritius AL, Rapola J, van Diggelen OP, Saarela J, Jalanko A, Peltonen L (2004) A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum Mol Genet 13:2893-2906.
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373:2055-2066.
Lim JA, Kakhlon O, Li L, Myerowitz R, Raben N (2015) Pompe disease: Shared and unshared features of lysosomal storage disorders. Rare Dis 3:e1068978.
Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U, Sheridan SD, Lucente D, Sims KB, Gusella JF, Sondhi D, Crystal RG, Reinhardt P, Sterneckert J, Schöler H, Haggarty SJ, et al. (2014) Human iPSC models of neuronal ceroid lipofuscinosis capture distinct e ff ects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet 23:2005-2022.
Lubs HA, Stevenson RE, Schwartz CE (2012) Fragile X and X-linked intellectual disability: four decades of discovery. Am J Hum Genet 90:579-590.
Nelakanti RV, Kooreman NG, Wu JC (2015) Teratoma formation: a tool for monitoring pluripotency in stem cell research. Curr Protoc Stem Cell Biol 32:4A 8 1-17.
Pasca SP, Panagiotakos G, Dolmetsch RE (2014) Generating human neurons in vitro and using them to understand neuropsychiatric disease. Ann Rev Neurosci 37:479-501.
Rossor MN, Fox NC, Mummery CJ, Schott JM, Warren JD (2010)e diagnosis of young-onset dementia. Lancet Neurol 9:793-806.
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, Lopez-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A (2012) Disease-speci fi c phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4:380-395.
Schmiedt ML, Blom T, Blom T, Kopra O, Wong A, von Schantz-Fant C, Ikonen E, Kuronen M, Jauhiainen M, Cooper JD, Jalanko A (2012) Cln5-de fi ciency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol Dis 46:19-29.
Sreedharan J, Brown RH, Jr. (2013) Amyotrophic lateral sclerosis: Problems and prospects. Ann Neurol 74:309-316.
Sugai K, Fukuzawa R, Shofuda T, Fukusumi H, Kawabata S, Nishiyama Y, Higuchi Y, Kawai K, Isoda M, Kanematsu D, Hashimoto-Tamaoki T, Kohyama J, Iwanami A, Suemizu H, Ikeda E, Matsumoto M, Kanemura Y, Nakamura M, Okano H (2016) Pathological classification of human iPSC-derived neural stem/progenitor cells towards safety assessment of transplantation therapy for CNS diseases. Mol Brain 9:85.
Trilck M, Hubner R, Seibler P, Klein C, Rolfs A, Frech MJ (2013) Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease speci fi c hallmarks. Orphanet J Rare Dis 8:144.
Uusi-Rauva K, Blom T, von Schantz-Fant C, Blom T, Jalanko A, Kyttala A (2017) Induced pluripotent stem cells derived from a CLN5 patient manifest phenotypic characteristics of neuronal ceroid lipofuscinoses. Int J Mol Sci 18:E955.
Virgo L, de Belleroche J (1995) Induction of the immediate early gene c-jun in human spinal cord in amyotrophic lateral sclerosis with concomitant loss of NMDA receptor NR-1 and glycine transporter mRNA. Brain Res 676:196-204.
Yamanaka S (2008) Pluripotency and nuclear reprogramming. Philos Trans R Soc Lond B Biol Sci 363:2079-2087.
Zhang Y, Schmid B, Nikolaisen NK, Rasmussen MA, Aldana BI, Agger M, Calloe K, Stummann TC, Larsen HM, Nielsen TT, Huang J, Xu F, Liu X, Bolund L, Meyer M, Bak LK, Waagepetersen HS, Luo Y, Nielsen JE; FReJA Consortium, et al. (2017) Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Rep 8:648-658.
Cindy E. McKinney, Ph.D., cmckinney@carolinas.vcom.edu.
10.4103/1673-5374.211180
*< class="emphasis_italic">Correspondence to: Cindy E. McKinney, Ph.D., cmckinney@carolinas.vcom.edu.

- 中国神经再生研究(英文版)的其它文章
- SoxC transcription factors in retinal development and regeneration
- Umbilical cord: an unlimited source of cells di ff erentiable towards dopaminergic neurons
- Targeting 14-3-3 adaptor protein-protein interactions to stimulate central nervous system repair
- RACK1 regulates neural development
- Schwann cell development, maturation and regeneration: a focus on classic and emerging intracellular signaling pathways
- BDNF pro-peptide: a novel synaptic modulator generated as an N-terminal fragment from the BDNF precursor by proteolytic processing