
致死性侏儒症Ⅰ型高发突变发生机制浅析
2017-07-21 09:41姜煜郭东炜郭奕斌
分子诊断与治疗杂志 2017年4期
姜煜郭东炜郭奕斌
综 述
致死性侏儒症Ⅰ型高发突变发生机制浅析
姜煜1郭东炜2郭奕斌1
致死性侏儒症(thanatophoric dysplasia,TD)是重型短肢畸形病中相对常见的致死性遗传性骨病,分为TD-I和TD-II 2型,它们都是由于FGFR3基因发生致死突变所致。TD-I型存在多种致病性突变,其中以c.742C〉T/p.R248C突变最为常见。本病为常染色体显性遗传(autosomal dominant,AD),杂合突变即可致死,但为何表型、基因型正常的父母会生出带有p.R248C突变的患胎并且是纯合突变的死胎?如果是新生突变,又为何会接连数胎生出同样的患胎?p.R248C高发突变的机制是什么?本文重点围绕这些问题,从“FGFR3(Fibroblast growth factor receptor 3)基因和FGFR3受体(Fibroblast growth factor receptor 3)的结构和功能,TD-I型p.R248C高发突变的发生机制,正常父母生出纯合致死突变的可能机制”几方面进行剖析,指出:①FGFR3基因及其受体蛋白结构和功能的特殊性是高发突变发生的物质基础;②位于IgⅡ和IgⅢ结构域连接区的氨基酸是极性很强的亲水氨基酸,很容易与带电离子结合而影响α螺旋结构,故易受外来理化因素的攻击、诱变而发生改变;③正常父母生出纯合致死突变,推测有两种可能:一种是夫妇一方的生殖腺已带有该高发突变,当胎儿从亲代遗传了一次突变后,只需在另一位点上再发生一次新生突变即可产生;另一种是夫妇双方的生殖腺都是该突变的嵌合体,当两者结合在一起,就有可能产生纯合突变。此外,还对未来发展趋势进行了展望。本文旨在探讨c.742C〉T/p.R248C高发突变和纯合突变发生的机制,进而为其今后的诊断和防治工作提供理论依据。
致死性侏儒症I型(TD-I);FGFR3基因;p.R248C;高发突变;发生机制
致死性侏儒症(thanatophoric dysplasia,TD,MIM187600)又称致死性骨发育不良,是新生儿致死性骨发育不良病中相对常见的一种,主要表现为肢体显著短小,巨颅,前额隆凸,脑部畸形,脊柱异常,肋骨极短,胸廓极度狭窄,四肢外旋或外展,呈蛙式体态[1-4],见图1、图2。本病为AD遗传,新生儿发生率国内报道约为1/40 000[3],国外报道为1/20 000~1/50 000[5]。患胎多半胎死腹中或于出生后不足24 h即因重症急性呼吸衰竭而夭折,但不同病例其病程进展会有差异[6-7],但一年存活率几乎为 0[8]。
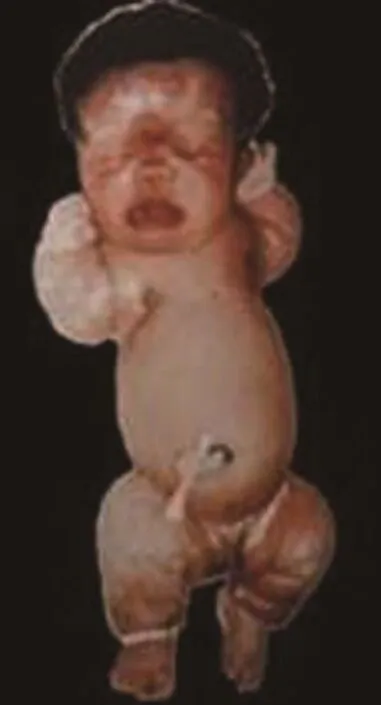
图1 TD-Ⅰ型Figure 1 TD typeⅠ[2]
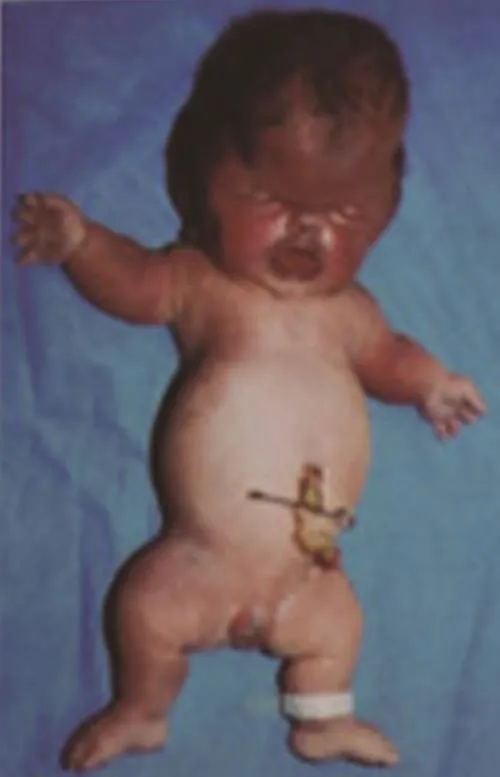
图2 TD-Ⅱ型[2]Figure 2 TD typeⅡ[2]
TD是由成纤维细胞生长因子受体3(Fibroblast growth factor receptor 3,FGFR3)基因发生不同突变所致,其中p.R248C错义突变为TD-Ⅰ最高发的致死突变,而TD-Ⅱ型多由p.K650E错义突变所引起。
TD-Ⅰ为AD遗传,杂合突变即可致死,但临床实践中,我们发现不少表型、基因型正常的父母会生出TD-Ⅰ患胎特别是p.R248C突变的患胎,如近几年,我们在10例TD-Ⅰ患胎中,就发现有6例是由p.R248C突变所致的死胎,其中纯合突变占了一半(3例),比例相当高。为何表型、基因型正常的父母会生出p.R248C的患胎特别是纯合突变的患胎?如果是新生突变,又为何会接二连三孕育出同样的患胎?是什么原因导致FGFR3受体蛋白的第248位的精氨酸(R)如此容易受到攻击?患胎的纯合突变又来自哪里?本文重点围绕这些问题,从“FGFR3基因和FGFR3受体的结构和功能,TD-Ⅰ型p.R248C高发突变的发生机制,正常父母生出纯合致死突变的可能机制”几方面进行剖析,以期为其今后的诊断和防治工作提供理论依据。
1 FGFR3基因和FGFR3受体的结构和功能
人FGFR3基因(OMIM:134934)定位于4p16.3,gDNA全长15 561 bp,有18个外显子和17个内含子。其中,第1外显子的全部和第2外显子的前半部分均不参与编码蛋白质,FGFR3跨膜区由第9外显子编码。FGFR3基因的序列在不同物种之间具有高度同源性。其开放阅读框(open reading frame,ORF)长13 328 bp,cDNA 2 421 bp,编码806个氨基酸(from RefSeq NM_000142),分子量为110 kDa~135 kDa。分子量的不同可能与其mRNA的剪接方式有关,功能上有重要意义的剪接主要发生在IgⅢ(图3)。作为发育调节跨膜受体之一的FGFR3,它能通过诱导骨成型蛋白质(bone morphogenetic protein,BMP)Ⅰ型受体的降解来调控骨骼的发育[9]。FGFR3属于免疫球蛋白样受体酪氨酸激酶(receptor tyrosine kinases,RTKs)家族,该家族还包括 FGFR 1、2、4。成纤维细胞生长因子(fibroblast-growth factors,FGFs)的靶细胞的表面广泛分布这些跨膜受体,并且能够介导FGFs的生物学活性。同族的跨膜受体的结构相似,均为跨膜受体[10-11]。FGFR3受体蛋白由胞内区、跨膜区和胞外区3部分组成,其中胞外区包含3个免疫球蛋白样功能域(IgⅠ,IgⅡ和IgⅢ)和一个肝素结合功能域(heparin-binding domain),IgⅠ和IgⅡ之间有一酸性区(acidic region,A)[12]。FGFRs只跨膜一次。膜内部分由一个内在激酶功能域(interkinase domain)、2个保守的酪氨酸激酶功能域(TK1,TK2)、近膜区和可发生自身磷酸化的C末端构成。FGFR3由18个外显子编码,7、8、9号外显子编码IgⅢ,其中7、8号外显子编码IgⅢb,7、9号外显子编码IgⅢc,多种FGFs可与之结合。FGFs是肝素结合生长因子(heparin binding growth factor,HBGF)家族的一员,在硫酸乙酰肝素蛋白多糖(heparan sulfate proteoglycans,HSPGs)协同下FGFs与受体(FGFRs)的IgⅡ及IgⅢ区结合之后,受体自身的酪氨酸残基磷酸化,受体二聚化而活化,酪氨酸残基磷酸化后和SH2信号分子相互结合,导致胞浆内的SH2结构域蛋白与含丝氨酸/苏氨酸蛋白激酶(MAPK,MAPKK和MAPKKK)产生磷酸化,从而激活核内的转录因子(signal transdusor and activator of transcription,STAT)和调控信号向细胞内传导[13-14],通过 PLC-γ 、Ras-MAPK、PI3K等途径传递信号[15]。FGFRs家族有多个成员,它们均参与调控长骨的生长,包括软骨基质的钙化,血管的入侵,软骨细胞的增生和成骨的形成等多个阶段[16]。最近,研究人员采用定量成像福斯特共振能量转移(quantitative imaging förster resonance energy transfer,QI-FRET)法对衍生自囊泡的血浆膜(plasma membrane)的膜蛋白的相互作用特性进行深入研究,发现RTKs在骨骼发育过程中起着极为重要的作用,从而影响了FGFR3的二聚化[17]。突变引起的FGFR3激活程度会极大影响软骨疾病表型的严重程度,其中引起TD的突变的激活程度最强。TD-Ⅰ型患者已发现12种不同突变(见HGMD数据库,http://www.hgmd.cf.ac.uk/ac/index.php),都会导致突变后的FGFR3功能持续地激活,导致FGFR3对骨骼生长的负向调节作用剧增,进而会导致患者骨骼生长发育的障碍。图3为已知人FGFR3突变分布及受体结构的图。
2 TD-Ⅰ型p.R248C高发突变的发生机制
临床上发现好多基因型和表型均正常的父母却生出了带有p.R248C突变的患胎。是什么原因导致FGFR3基因发生该致死突变?为何第248位的精氨酸(R)如此容易受到攻击?
虽然目前发现有12种突变(多数为单个氨基酸突变成Cys)都可以引起TD-Ⅰ,比如集中在FGFR3蛋白胞外区IgⅡ和IgⅢ结合域连接区的第248位精氨酸(R)、第249位丝氨酸(S),近膜区的第370位甘氨酸(G)、371位丝氨酸(S)、373位酪氨酸(Y)都是被半胱氨酸(C)取代即R248C、S249C、G370C、S371C、Y373C,此外,还有 K650M以及终止密码子(TGA)发生的突变(X807R、X807C、X807G、X807L、X807S、X807W,突变使羧基端延长141个氨基酸)均可导致TD-Ⅰ型的发生[19]。但最常见的仍是发生在第248位的p.R248C错义突变,已报道的由此突变引起的TD-Ⅰ病例已超过100例,约占所有突变的55%[20]。

图3 人FGFR3受体结构及已知突变的分布[12,18]Figure 3 The structure of human FGRG3 receptor and the distribution of known mutations[12,18]
从FGFR3的分子结构可知跨膜区(E9)为保守的疏水性区段(AGSVYAGILSYGVGFFLFILVVAAVTLC),2个酪氨酸激酶活性区位于细胞的内段(即胞内区)。此两者之间有一插入序列(GPLYVLVEYAAKGNLRE)将其分开,是FGFR3受体的高度保守部分[21]。当配体和胞外区结合后,受体就会形成同源或异源的二聚体(dimer)[5],导致酪氨酸激酶在细胞内段的活化,催化细胞的酪氨酸残基发生磷酸化,也致使酪氨酸残基在受体靶蛋白上磷酸化,使得级联反应在细胞内发生,使DNA最终合成导致细胞分裂[22]。
在小鼠模型研究中,加入酪氨酸激酶抑制剂可改善与FGFR3受体相关的侏儒症的症状[23]。高度疏水的跨膜区被该跨膜区的突变氨基酸所影响,在缺乏FGFs配体(ligand)时,此突变导致一个α螺旋形成,可以激活受体(receptor)。功能获得性FGFR3基因突变常导致骨骼发育不良类的疾病,增高了受体酪氨酸磷酸化的水平,导致了非配体(unligand)的依赖性及组成性的激活(constitutive activation)。由于FGFR3基因突变的类型和位置不同,所以产生异常的氢键、二硫键,使FGFR3单体异常聚合成二聚体,或者激酶区构象改变形成激活环,这些可能都是受体激活的原因[24]。位于IgⅡ和IgⅢ结构域的连接区,前后几个氨基酸(E,R,P,H,R,P)都是极性很强的亲水氨基酸或亚氨基酸,很容易与带电离子结合而影响原来的α螺旋结构,故易受外来理化因素(电离射线和多种多样的化学药物)的攻击而发生改变。一旦突变成Cys,就有可能形成二硫键,从而导致IgⅡ和IgⅢ结构的改变,致使稳定性较跨膜区和胞内区(羧基端侧)差。
研究发现,TD-Ⅰ的FGFR3基因的c.742C〉T/p.R248C突变可以引起c-fos基因(为一种即刻早期基因,其表达产物Fos蛋白与靶基因的特异序列结合,调节其转录水平发挥信使作用)的转录活性比野生型增加4倍[25]。而对照组p.R248A突变体与野生型则无明显差别。c-fos基因高度保守,是编码核蛋白的基因,Fos内磷酸化蛋白是c-fos基因的表达产物,此产物的结构特点:DNA结合区域由碱性氨基酸构成。附近的亮氨酸残基是由7个氨基酸组成的,亦称之为亮氨酸拉链(LZ),它与另一端的并列的亮氨酸形成第一个α螺旋,含有该结构的其他蛋白和亮氨酸残基并列形成二聚体,然后与靶基因的碱性激活因子-1(AP-1)结合,调节靶基因的表达[17]。
研究还发现p.R248C突变的FGFR3嵌合受体可以引起NIH3T3细胞(小鼠胚胎成纤维细胞,是疏松结缔组织的主要细胞成分)的转化,而NIH3T3细胞的转化不受p.R248A突变的嵌合受体影响,可以说明功能获得性方式引起TD-Ⅰ型。引入Cys可以导致通过二硫键在受体之间形成二聚体,影响此区域的α螺旋结构,导致受体非配体依赖性的组成性活化[23]。当孕妇在孕期由于患病而用药时或在孕早期接触了一些电离射线,由于带电离子容易与p.248的精氨酸(R)等极性氨基酸结合而发生突变,故当接触或服用了含有带电离子的药物等理化诱变因子时就容易导致p.248位的R的基团发生改变而引起致死突变,这一点与同是AD遗传的成骨不全症I型(或Ⅱ型)的COL1A1/COL1A2基因的突变机制类似[26]。这也许可以部分解释为何表型和基因型正常的父母会生出带有p.R248C新生致死突变的患胎。
3 正常父母生出纯合致死突变的可能机制
如果只引起一个位点(特别是突变热点)的新生突变,并不难理解。令人费解的是基因型(来自外周血组织)和表型均正常的父母为何会那么凑巧在同一位点发生相同突变而生出纯合突变的患胎?Takagi等[27]2012年报道:一些非典型软骨发育不全(achondroplasia,ACH)症状的患儿存在着p.R248C突变的体细胞嵌合体。因此,我们推测生出纯合突变的患胎有2种可能:一种是夫妇一方的生殖腺(如父亲的精巢或母亲的卵巢)已经带有该高发突变,当胎儿从父或母遗传了一次突变后,只需在另一个高发等位基因位点上再发生一次新生突变即可产生纯合突变(类似于肿瘤发生的二次突变学说);另一种情况可能是夫妇双方的生殖腺(精巢和卵巢)都是该突变的嵌合体,因此都有可能产生带有这一突变的生殖细胞(精子和卵子)。当两者结合在一起,就有可能产生纯合突变。在这种情况下,正常的父母连续数胎生出TD-Ⅰ患胎就不难理解了。这种情况与成骨不全患胎的父母的生殖腺存在COL1A1/COL1A2突变嵌合体类似。对于成骨不全Ⅱ型(胎儿致死型)来说,一般是见不到家族史的,因此再次生育成骨不全型Ⅱ型(osteogenesis imperfecta typeⅡ,OI-Ⅱ)型患胎的可能性很小。但有文献报道,有些家系却多次孕育致死的OI-Ⅱ型患胎,经研究,原来是与夫妇的生殖腺存在突变嵌合体有关[28]。
此外,正常表型的父母产生的TD-Ⅰ型患胎可能与父亲的年龄及精液中存在突变的嵌合体的关系更为密切。据文献报道,大多数AD遗传的新突变基因都发生在父亲的等位基因[29],这可能是由于随着父亲年龄增长,精子细胞的不断产生,其发生突变的机会也在不断增加。有研究表明突变是通过异常的侵袭过程完成的[18-19,30-31]。当然也不能排除患胎的突变是来自高龄母亲的嵌合体卵巢。
4 展望
致死性侏儒症胎儿多自发流产或人工引产[32],少数出生的患胎也多于出生后24 h内死亡。虽然有个别TD-Ⅰ型病例(带有p.R248C突变)可以存活下来,症状有点类似于ACH,但研究表明该病例的体细胞存在着嵌合体,而且正常细胞的比例比较大[27]。该病至今仍无行之有效的根治疗法,但 Yamashita等[33]2014 年报道,他们用“他汀类”(statin)治疗剂在TD-Ⅰ的软骨分化和ACH多能干细胞(induced pluripotent stem cell,iPSCs)的诱导过程中能矫正初级软骨(degraded cartilage)的形成。用“他汀类”治疗的ACH老鼠模型能明显促进骨的生长的恢复。这些结果提示,“他汀类”能代替TD-Ⅰ和ACH婴幼患儿的医学治疗[33]。但这些研究仍只停留在动物模型阶段,因此目前应对该病的最好方法就是预防突变的发生以及对高危妊娠进行产前诊断以防止患儿出生,或采取胚胎植入前遗传学诊断(preimplantation genetic diagnosis,PGD)的手段预防患胎的孕育。而揭示TD的发病机制尤其是高发突变的发生机制将有助于进行有效的防患。
然而,对于TD-Ⅰ型p.R248C高发突变的机制至今仍未被完全阐明。对于正常父母接连生出患胎的可能原因——源自生殖腺嵌合体,目前也只是处于理论推测阶段。因此科研人员今后可朝这方面进行深入研究以获得更有说服力的实验证据。
而要彻底揭示其发生机制,还有必要对患胎父母的多种组织进行深入研究以及对他们进行详细随访,了解其家族史及孕前、孕期的生活、工作环境和生活习惯等,或许患胎的自发突变并非遗传自父母的一方或双方,而是由于后天接触某类化学或生物或物理因素而诱发生殖细胞发生基因突变,关于这一点,前面已有阐述,但理、化、生诱变因素具体是哪些?它们的原子、电子结构如何?诱变机制又是怎样?动物模型又该如何建立?则有待科研人员今后花费大量的精力、人力和财力去探究。
另外,随着各种罕见病例的增多和研究的不断深入,已经逐步阐明同是FGFR3基因突变引起的骨骼发育异常病,只要突变部位、突变类型以及作用于FGFR3受体蛋白的结构域存在不同,就可导致患者(或患胎)的临床表型存在很大差异,从生长发育、身高体重、智力水平、寿命基本正常如季肋发育不全(Hypochondroplasia,HCH)到个头矮小、骨骼畸形、O型腿、头颅异常、寿命较短如软骨发育不全(ACH),再到胎死腹中或产下即死如致死性侏儒症(TD-Ⅰ或TD-Ⅱ)。此外,还阐明即使带有相同突变,只要突变等位基因的杂合或纯合状态不同,或患者体细胞或生殖腺细胞有无存在嵌合体,也会产生差别明显的临床症状,比如杂合的p.G380R突变仅仅引起ACH,但纯合的p.G380R突变却会导致胎儿围产期死亡或产后不久即夭折,类似于TD病;而体细胞存在p.R248C杂合致死突变的嵌合体患者,却能像ACH患者一样正常存活,甚至生儿育女。可见FGFR3基因的突变存在着明显的剂量效应。那么,多大剂量、多大比例的p.G380R突变才会导致类似于TD的症状出现?又是多大剂量、多大比例的p.R248C突变才会表现类似ACH呢?这些发生机制都有待于今后采用更先进、更精确的技术以及联合采用各种组学的方法进行深入的研究。
[1]Warman ML,Cormier-Daire V,Hall C,et al.Nosology and classification of genetic skeletal disorders:2010 revision[J].Am J Med Genet A,2011,155A(5):943-968.
[2]Kenneth Lyons Jones.SMITH人类先天性畸形图谱——分类、判定标准与遗传咨询[M].傅松滨,译.北京:人民卫生出版社,2007:377-378.
[3]刘权章.临床遗传学彩色图谱[M].2版,北京:人民卫生出版社,2006:319-320.
[4]Itoh K,Pooh R,Kanemura Y,et al.Brain malformation with loss of normal FGFR3 expression in thanatophoric dysplasia type I[J].Neuropathology,2013,33(6):663-666.
[5]Del Piccolo N,Placone J,Hristova K.Effect of thanatophoric dysplasia type I mutations on FGFR3 dimerization[J].Biophys J,2015,108(2):272-278.
[6]Chen CP,Chern SR,Shih JC,et al.Prenatal diagnosis and genetic analysis of type I and typeⅡthanatophoric dysplasia[J].Prenat Diagn,2001,21(2):89-95.
[7]de Souza Cambraia VD,Rezende MA,Roquetto Gomes KM.Severe acute respiratory failure caused by thanatophoric dysplasia.The report of two cases with different clinical developments[J].Pediatric Pulmonology,2016,51(SI):S60-S61,Suppl:42.
[8]Monti E,Mottes M,Fraschini P,et al.Current and emerging treatments for the management of osteogenesis imperfecta[J].Ther Clin Risk Manag,2010,6:367-381.
[9]Qi H,Jin M,Duan Y,et al.FGFR3 induces degradation of BMP type I receptor to regulate skeletal development[J].Biochim Biophys Acta,2014,1843(7):1237-1247.
[10]Sarabipour S,Hristova K.Mechanism of FGF receptor dimerization and activation[J].Nat Commun,2016,7:10262.
[11]Moldrich EX,Mezzera C,Holmes WM,et al.Fgfr3 regulates development of the caudal telencephalon[J].Dev Dyn,2011,240(6):1586-1599.
[12]Foldynova-Trantirkova S,Wilcox WR,Krejci P.Sixteen years and counting:the current understanding of fibroblast growth factor receptor 3(FGFR3)signaling in skeletal dysplasias[J].Hum Mutat,2012,33(1):29-41.
[13]Krejci P,Salazar L,Kashiwada TA,et al.Analysis of STAT1 activation by six FGFR3 mutants associated with skeletal dysplasia undermines dominant role of STAT1 in FGFR3 signaling in cartilage[J].Plos One,2008,3(12):e3961.
[14]Harada D,Yamanaka Y,Ueda K,et al.Sustained phosphorylation of mutated FGFR3 is a crucial feature Of genetic dwarfism and induces apoptosis in the ATDC5 chondrogenic cell line via PLCy-activated STAT1[J].Bone,2007,41(2):273-281.
[15]Powers CJ,McLeskey SW,Wellstein A.Fibroblast growth factors,their receptors and signaling[J].Endoc Relat Cancer,2000,7(3):165-197.
[16]Ibrahimi OA,Zhang F,Eliseenkova AV,et al.Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through Enhancement of FGF binding affinity[J].Hum Mol Genet,2004,13(1):69-78.
[17]Sarabipour S,Del Piccolo N,Hristova K.Characterization of membrane protein interactions in plasma membrane derived vesicles with quantitative imaging forsterresonanceenergytransfer[J].AccChem Res,2015,48(8):2262-2269.
[18]张晔.与FGFR3基因有关的遗传性侏儒研究进展[J].中国优生与遗传杂志,2000,8(1):1-3.
[19]Ornitz DM,Marie PJ.FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease[J].Genes Dev,2002,16(12):1446-1465.
[20]Passos-Buena MR,Wilcox WR,Jabs EW,et al.Clinical spectrum of fibroblast growth factor receptor mutations[J].Hum Mutat,1999,14(2):115-125.
[21]Jantip J,Tanthanuch M,Kanngurn S,et al.Mutations of fibroblast growth factor receptor 3 gene(FGFR3)in transitional cell carcinoma of urinary bladder in Thai patients[Revision-2a][J].J Med Assoc Thai,2013,96(8):976-983.
[22]Kwabi-addo B,Ropiquet F,Giri D,et al.Alternative splicing of fibroblast growth factor receptors in human prostate cancer[J].Prostate,2001,46(2):163-172.
[23]Komla-Ebri D,Dambroise E,Kramer I,et al.Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model[J].Journal of clinical investigation,2016,126(5):1871-1884.
[24]Ikegami D,Iwai T,Ryo S,et al.Identification of small molecular compounds and fabrication of its aqueous solution by laser-ablation,expanding primordial cartilage[J].Osteoarthritis Cartilage,2011,19(2):233-241.
[25]d′Avis PY,Roberson SC,Meyer AN,et al.Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type 1[J].Cell Growth&Diff,1998,9(1):71-78.
[26]李荣,郭源平,潘敬新,等.成骨不全I型家系的基因检测和COL1A2基因新突变的致病性鉴定[J].遗传,2015,37(1):41-47.
[27]Takagi M,Kaneko-Schmitt S,Suzumori N,et al.Atypical achondroplasia due to somatic mosaicism for thecommon thanatophoric dysplasiamutation R248C[J].Am J Med Genet A,2012,158A(1):247-250.
[28]Morgan JA,Marcus PS.Prenatal diagnosis and management of intrauterine fractur[J].Obstetrical&Gynecological Survey,2010,65(4):249-259.
[29]Gibbs L,Legeai-Mallet L.FGFR3 intracellular mutations induce tyrosine phosphorylation in the Golgi and defective glycosylation[J].Biochim Biophys Acta,2007,1773(4):502-512.
[30]Su N,Jin M,Chen L.Role of FGF/FGFR signaling in skeletal development and homeostasis:learning from mouse models[J].Bone Res,2014,2:14003.
[31]Monsonego Ornan E,Adar R,Feferman T,et al.The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation[J].Mol Cell Biol,2000,20(2):516-522.
[32]Dias AT,Zanardo ÉA,Dutra RL,et al.Post-mortem cytogenomic investigations in patients with congenital malformations[J].Exp Mol Pathol,2016,101(1):116-123.
[33]Yamashita A,Morioka M,Kishi H,et al.Statin treatment rescues FGFR3 skeletal dysplasia phenotypes[J].Nature,2014,513(7519):507-511.
Analysis of the mechanism of high incidence of thanatophoric dysplasia type I
JIANG Yu1,GUO Dongwei2,GUO Yibin1
(1.Department of Medical Genetics,Zhongshan School of Medicine,SUN Yat-sen University,Guangzhou,Guangdong,China,510080;2.Clinical Medicine,Medical College,Xiamen University,Xiamen,Fujian,China 361102)
Thanatophoric dysplasia(TD),divided into TD type I and TD type II,is a genetic bone disease caused by the fatal mutation of FGFR3 gene.It is relatively common lethal hereditary osteopathy among severe short limb malformations.TD-I type has many kinds of pathogenic mutations,of which c.742C〉T/p.R248C mutation is the most common.This disease is an autosomal dominant(AD)genetic disease which means heterozygous mutations can be lethal.However,why some parents with normal genotypes and phenotypes can give birth to a stillbirth who carried the homozygous mutation p.R248C?If it is a novel mutation,why the successive fetuses with the same mutation were born?What is the high incidence mechanism of p.R248C mutation?This paper focus mainly around these problems,from“The structure and function of FGFR3 gene and FGFR3 receptor;The pathogenetic mechanism of high incidence of p.R248C in TD type I;The possible mechanisms of normal parents producing homozygous lethal mutation”,several aspects are analyzed and pointed out:①FGFR3 gene and the structure and function of its receptor protein is the material base of high frequentmutation;②Some amino acids,located in the connected region of IgⅡand IgⅢdomain,are strong polar hydrophilic amino acids,which tend to bind charged ions to influence the alpha helical structure,therefore,it is easy to be affected by external physical and chemical factors;③Normal parents give birth to fetus with homozygous lethal mutation,there are 2 possibilities:one is the reproductive gonad of one of the parents has carried the hot mutation,and the fetus inherited the hot mutation,under these circumstances,as long as a same mutation happen in its allele,which will lead to the generation of homozygous mutation;another is the reproductive glands of couples are the mutant chimeras,when the two are combined together,it is possible to generate homozygous mutation.In addition,the future development trend is prospected.The purpose of this paper is to explore the high-risk mutation and homozygous mutation mechanism of c.742C〉T/p.R248C,and to provide theoretical basis for the diagnosis and prevention and control work in future.
Thanatophoric dysplasia type I(TD-I);FGFR3 gene;p.R248C;High mutation;Pathogenetic mechanism
闽粤合作科研基金(71010025)
1.中山大学中山医学院医学遗传室,广东,广州510080
2.厦门大学医学院临床医学专业,福建,厦门361102
郭奕斌,E-mail:aguoabin@qq.com
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
科学与社会(2021年3期)2021-12-02
中国生殖健康(2020年4期)2021-01-18
生殖医学杂志(2019年11期)2019-02-15
中成药(2018年12期)2018-12-29
中国生殖健康(2018年4期)2018-11-06
天然产物研究与开发(2018年1期)2018-02-02
中成药(2018年1期)2018-02-02
发明与创新·中学生(2017年6期)2017-06-12
方圆(2016年15期)2016-09-14
