
自身免疫性癫痫
2017-07-19 13:48陈艳艳卓综述林卫红审校
中风与神经疾病杂志 2017年6期
陈艳艳, 王 卓综述, 林卫红审校
自身免疫性癫痫
陈艳艳, 王 卓综述, 林卫红审校
随着自身免疫性中枢神经系统疾病(central nervous system,CNS)研究的快速发展,癫痫与免疫的关系也渐成热点,目前已证实免疫炎症反应与癫痫密切相关[1],但自身免疫性癫痫(autoimmune epilepsy,AE)尚无标准的定义、诊断及治疗的管理指南。本文从AE的流行病学、发病机制、临床特点、诊断、治疗及预后做一综述,旨在提高神经科医生对于AE的认识。
1 流行病学
癫痫的患病率约为0.4%,而AE的患病率仍不清楚,对于检出抗神经元抗体、合并有自身免疫性疾病(autoimmune diseases,AD)或免疫调节治疗有效的癫痫患者均怀疑为AE[2]。有研究显示高达11%~16%的癫痫患者检出抗神经元抗体[3],儿童新发癫痫队列研究中检出率为9.7%[4]。大样本(n=2,518,034)调查显示,17.5%的癫痫患者同时患有AD,而AD并发癫痫的风险增加了4倍,儿童中更是增加了5倍[2]。一项AD与癫痫关系的Meta分析也得出明显相关的结论,癫痫患AD的风险增加2.6倍,而AD患癫痫的风险增加2.7倍,年龄<20岁的患者风险更高[5]。
2 发病机制
已知细胞免疫、体液免疫和细胞因子均参与癫痫的免疫致病过程[1],但抗神经元抗体是通过何种途径介导癫痫发病尚在研究中。根据分布范围将抗神经元抗体分为Ⅰ型抗体(抗神经元细胞内抗原抗体)和Ⅱ型抗体(抗神经元表面抗原抗体),后者明显多于前者[6]。目前认为Ⅰ型抗体本身无致病性,是通过CD4+/CD8+T细胞介导的细胞毒性反应产生致病作用,包括抗ANNA-1(Hu)抗体、抗Ma1/Ma2(Ta)抗体、抗GAD抗体、抗CRMP-5抗体、抗AMPH抗体,与恶性肿瘤相关,对免疫治疗效果差[7];而Ⅱ型抗体被认为是致病性自身抗体,可能的发病机制是改变靶抗原的数量或功能起到直接致病作用,包括抗VGKC复合物抗体(抗LGi1抗体/抗CASPR2抗体/其他)、抗NMDAR抗体、抗AMPAR抗体、抗GABA(a/b)抗体、抗mGLuR5抗体等,对免疫治疗效果相对较好;还有一些抗体的发病机制不明确,怀疑是与其他抗神经元抗体共同作用,包括抗gAChR抗体、抗VGCCs(N/P/Q)抗体、抗DPPX抗体[8]。各种抗神经元抗体的可能的致病机制(见表1)[7~9]。
3 临床表现
3.1 临床特征 癫痫发作是AE的最突出症状,多表现为部分性发作并快速进展为癫痫持续状态(SE)或严重的簇状发作[10],且药物难治性癫痫发作多见,可能同时伴有其他神经功能症状(如认知障碍、运动障碍、自主神经功能障碍和意识障碍等)和精神症状(如个性和行为改变、幻觉、焦虑、抑郁等)[3,11]。根据可能的病因将AE分为自身免疫性脑炎相关癫痫、AD相关癫痫、抗神经元抗体阳性或免疫治疗有效的难治性癫痫和癫痫综合征。
3.1.1 自身免疫性脑炎相关癫痫 自身免疫性脑炎相关癫痫以癫痫发作、认知功能下降、精神行为异常以及不自主运动为主要临床表现[3]。癫痫发作可为首发症状或出现于病程任何时期,复杂部分性发作、全面性发作、癫痫持续状态、非惊厥性癫痫持续状态(NCSE)、肌阵挛等均有报道[7]。 不同抗体类型的AE可能有其特征性的运动障碍,如抗LGi1脑炎性癫痫特征性的表现为面-臂肌张力障碍样发作(FBDS);抗NMDAR脑炎性癫痫可表现为口面部不自主运动;抗Ma2和CRMP-5相关癫痫可表现为帕金森病症状;抗GAD65相关癫痫可表现为僵人综合征(SMS)。自身免疫性脑炎相关癫痫的临床特点(见表2)[6,10,12~14]。
3.1.2 AD相关癫痫 AD临床表现多样性,可涉及多个脏器。多数AD并发癫痫是因为出现了相关的脑病(如狼疮脑病、桥本氏脑病等)或本身为神经系统免疫性疾病(如多发性硬化),癫痫发作可为首发症状、惟一症状或突出症状,发作类型包括部分性发作、全面性强直-阵挛发作、肌阵挛等,可同时伴有神经症状(认知障碍、肢体活动障碍、言语障碍、自主神经功能障碍等)和精神症状(谵妄、幻觉、抑郁等)[11]。
3.1.3 抗神经元抗体阳性或免疫治疗有效的难治性癫痫和癫痫综合征 一部分孤立性癫痫发作的患者中同样发现了抗神经元抗体,多见于隐源性局灶性癫痫(12.7%)和伴海马硬化的癫痫(23%)[3],且与上述抗体类型相同。儿童期常见的West综合征、获得性失语综合征(LKS)等被认为发病与免疫机制相关,以免疫治疗为一线治疗,但尚无证据证明与抗神经元抗体相关,是否为自身免疫性癫痫有待进一步研究。
3.2 辅助检查
3.2.1 实验室检查 血清和/或脑脊液抗神经元抗体阳性是诊断AE的一个重要参考标准。脑脊液蛋白增高是一种非特异性CNS炎症的标志,特别是IgG定量和合成指数的增高更有意义;寡克隆带阳性虽然对诊断无特异性,但在自身免疫性脑炎相关癫痫中普遍存在,尤其是抗NMDAR抗体阳性的患者中[15]。脑脊液新喋呤是急性和活动性CNS炎症的敏感指标,但不能区分原发性与继发性炎症反应,且没有被广泛应用[16]。CNS感染有多种细胞因子和趋化因子的参与,有可能成为新的脑脊液炎性标志物,目前仍在研究中。也有研究认为非抗神经元的特异性自身抗体(包括抗核抗体、抗心磷脂抗体、抗磷脂抗体、甲状腺过氧化物酶抗体等)在难治性癫痫患者中阳性率明显增高,并认为与AE有相关性,故有学者也把它们作为怀疑AE的一个参考指标[12]。
3.2.2 脑电图检查 脑电图在AE和其他癫痫患者中无统计学差异,但对于影像学及脑脊液检查正常的患者,一些相对特异性的脑电图表现可作为指导抗神经元抗体检测的指标[13]。研究显示脑电图中出现NCSE、额叶间断节律性δ波活动(FIRDA)、δ刷、伴有δ暴发的阵发性θ/δ慢活动可能与AE有关[13]。
3.2.3 影像学检查 大多数AE患者的头部MRI检查是正常的,尤其在发病早期。AE的MRI可出现颞叶内侧异常信号,也有报道皮质和皮质下、基底节、小脑等部位出现异常信号[10],晚期可出现海马萎缩。若临床高度怀疑,应考虑重复MRI检查。PET-CT扫描与MRI检查可以相互补充。FDG-PET异常与抗神经元抗体滴度之间的相关性已有报道,颞叶内侧高代谢更多见于Ⅰ型抗体并可能出现于临床症状和MRI异常之前。一般来说,FDG-PET比MRI更敏感,尤其是非颞叶(如脑干、大脑皮质或小脑等)区域,这些部位的代谢异常可能无临床症状[17]。
3.2.4 组织病理学检查 对抗神经元抗体阳性的患者进行组织活检及病理学检查,结果显示不同抗体类型均出现了组织病理学的炎症变化[14]。
4 诊 断
目前尚无AE的诊断标准,一般把血清/脑脊液抗神经元抗体阳性或免疫治疗有效的癫痫患者诊断为AE[3]。怀疑AE的诊断要点如下[7,12,14]:(1)临床特点:特征性的发作性症状,如:FBDS;SE(包括NCSE)或癫痫成簇状发作;不明原因的急性或亚急性癫痫发作(发作频率最长间隔<3 m),多为局灶性发作、继发性全面强直-阵挛发作;抗癫痫药物耐药;伴随症状:脑病,运动障碍,神经精神症状,认知和记忆障碍;前驱病毒感染;AD病史(个人或直系亲属);新发或既往肿瘤病史,尤其是与自身免疫性癫痫相关的肿瘤;免疫治疗(糖皮质激素、丙种球蛋白、利妥昔单抗等)有效;不能用其他病因(如代谢、感染、颅内肿瘤、脑血管病、外伤等)解释。(2)辅助检查:脑脊液蛋白增高、白细胞数增多、寡克隆带阳性、IgG合成率增加等符合CNS炎性反应的特点;头部MRI扫描 FLAIR/T2加权相显示颞叶或脑实质高信号;FDG-PET显示高代谢;脑电图出现慢波、癫痫波、FIRDA或“δ刷”;AD的血清学标记如抗核抗体或甲状腺过氧化物酶抗体阳性;组织病理活检发现炎性细胞浸润;血清和/或脑脊液抗神经元抗体阳性。
5 治疗及预后
传统抗癫痫药物治疗AE疗效不佳,免疫治疗在AE患者中有效率达60%~80%[18],年龄较大、Ⅱ型抗体对于早期免疫治疗的效果更佳,但MRI特点、发作类型、急性免疫治疗方案的选择或者慢性免疫调节治疗等对治疗结果无显著差异[6]。对于自身免疫性脑炎相关癫痫进行免疫治疗基本没有争议,但药物难治性癫痫患者何时启动免疫治疗未达成共识。目前尚无治疗指南,但均以“3M”为指导原则,即最大可逆性(至少发作频率减少>50%)(Maximum reversibility)、维持可逆性变化(Maintenance of reversibility)和最小的治疗剂量(Minimal therapeutic dose)[12]。目前公认的基本治疗方案(见表3)。
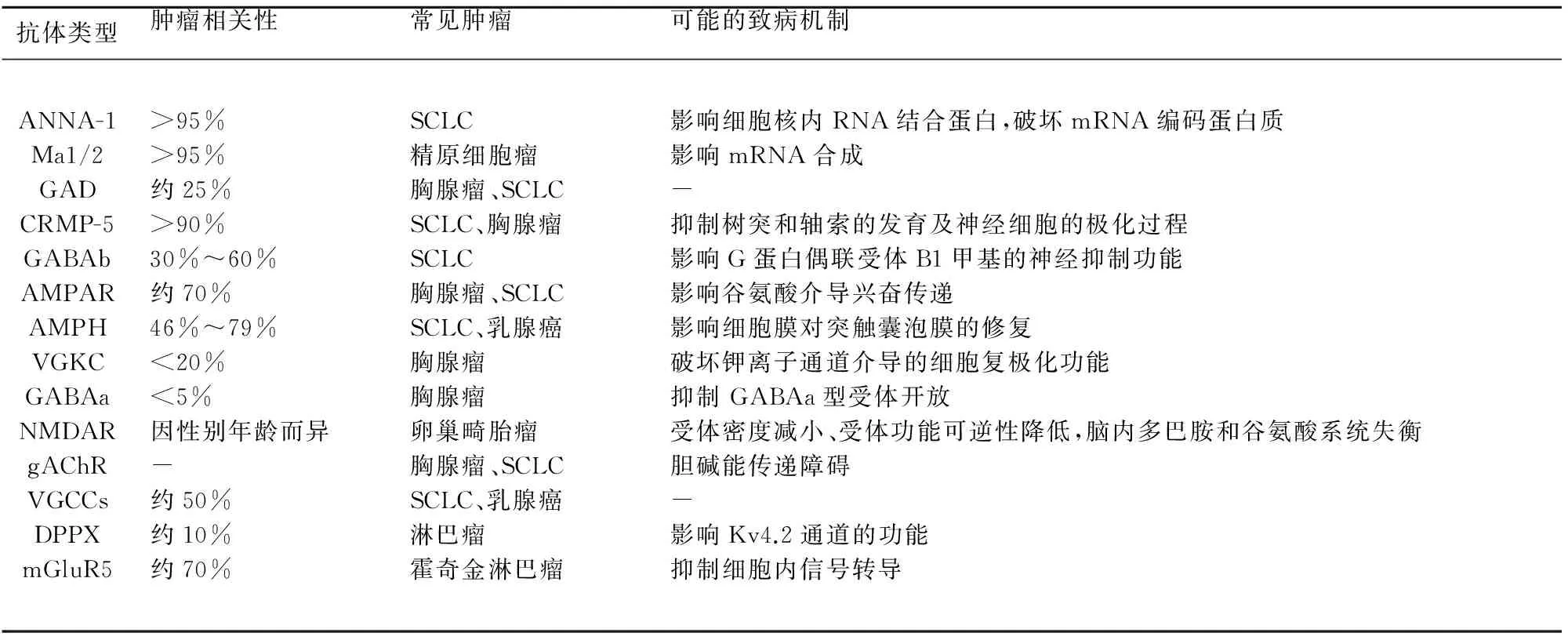
表1 各种抗神经元抗体可能的致病机制
注:-:尚不明确;SCLC:小细胞肺癌

表2 自身免疫性脑炎相关癫痫的临床特点
注:LE:边缘叶脑炎;FAD:家族性自主神经功能异常;OMS:眼阵挛-肌阵挛综合征; LEMS:Lambert-Eaton肌无力综合征; PLED:周期性一侧癫痫样放电

表3 AE的治疗
注:IVIG:静脉注射丙种球蛋白;MMF:麦考酚吗乙酯;i.v.:静脉注射;po.:口服 ;*:国内未批准用于治疗AE
选择一线或二线治疗需考虑多方面因素,包括适应证、安全性、不良反应、疗效等,不同的抗神经元抗体类型有不同的复发风险[14],任何治疗都需个体化选择。一线治疗也作为诊断性治疗在应用。国外研究发现利妥昔单抗作为二线治疗对所有抗体类型均受益,国内尚未批准其用于治疗AE,需综合评估病情并充分知情同意[9]。建议未发现肿瘤的Ⅰ型抗体阳性患者进行肿瘤筛查,每6个月一次,持续数年,以便早期发现潜在的恶性肿瘤并进行治疗[14]。在抗神经元抗体阳性的难治性癫痫患者中出现免疫治疗抵抗可以考虑手术治疗,手术原则应遵循术前评估的结果[19],疗效不确定。在整个治疗过程中抗癫痫药物仍需继续使用,免疫治疗有效的患者亦需要至少一种抗癫痫药物维持治疗。AE患者发生SE一般需用麻醉药物终止发作。免疫治疗维持的时间尚缺乏足够的证据,一般免疫治疗后病情稳定两年可考虑停用[12]。国内专家建议激素治疗维持6 m,如果口服免疫抑制剂要至少1 y[9],而Mckeon认为免疫治疗必须维持3~5 y才能停止[20]。治疗有效的关键是症状发作缓解、认知能力改善等。改良Ranking量表可作为治疗成功的评价标准,传统癫痫治疗有效(减少癫痫发作>50%)的评价标准也适用于AE[14]。
6 小结和展望
越来越多的证据支持AE的早期诊断和早期免疫调节治疗对改善患者的预后有积极的作用。由于目前无统一的诊断标准,又缺乏有效的需要检测抗神经元抗体的评价指标,患者是否受益于发病初期抗神经元抗体的筛查仍需进一步开展相关研究。缺乏大样本、多中心、随机对照试验,缺乏单独免疫治疗和抗癫痫药物治疗的比较研究,很难让临床医生直接放弃经验性抗癫痫药物治疗而首选免疫治疗。未来的研究应根据患者的临床表现和抗神经元抗体类型确定理想的免疫调节治疗方案。因此,AE的研究工作任重而道远。
[1]Vezzani A,Fujinami RS,White HS,et al.Infections,inflammation and epilepsy[J].Acta Neuropathol,2016,131(2):211-234.
[2]Ong MS,Kohane IS,Cai T,et al.Population-level evidence for an autoimmune etiology of epilepsy[J].JAMA Neurol,2014,71(5):569-574.
[3]Brenner T,Sills G,Hart Y,et al.Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy[J].Epilepsia,2013,54(6):1028-1035.
[4]Suleiman J,Wright S,Gill D,et al.Autoantibodies to neuronal antigens in children with new-onset seizures classified according to the revised ILAE organization of seizures and epilepsies[J].Epilepsia,2013,54(12):2091-2100.
[5]Zhang L,Qi Si,Zou XY.Association between epilepsy and systemic autoimmune diseases:a meta-analysis[J].Seizure,2016,41:160-166.
[6]Dubey D,Farzal,Z,Hays R,et al.Evaluation of positive and negative predictors of seizure outcomes among patients with immune-mediated epilepsy:a meta-analysis[J].Ther Adv Neurol Disord,2016,9(5):369-377.
[7]McKeon A,Pittock SJ.Paraneoplastic encephalomyelopathies:pathology and mechanisms[J].Acta Neuropathol,2011,122(4):381-400.
[8]Boronat A,Gelfand JM,Gresa-Arribas N,et al.Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6,a subunit of Kv4.2 potassium channels[J].Ann Neurol,2013,73(1):120-128.
[9]中华医学会神经病学分会.中国自身免疫性脑炎诊治专家共识[J].中华神经科杂志,2017,50(2):91-98.
[10]Hacohen Y,Wright S,Waters P,et al.Paediatric autoimmune encephalopathies:clinical features,laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens[J].J Neurol Neurosurg Psychiatry,2013,84(7):748-755.
[11]Vincent A,Crino PB.Systemic and neurologic autoimmune disorders associated with seizures or epilepsy[J].Epilepsia,2011,52(Suppl 3):12-17.
[12]Toledano M,Pittock SJ.Autoimmune epilepsy[J].Semin Neurol,2015,35(3):245-258.
[13]Baysal-Kirac L,Tuzun E,Altindag E,et al.Are there any specific EEG findings in autoimmune epilepsies[J].Clin EEG Neurosci,2016,47(3):224-234.
[14]Bien CG.Value of autoantibodies for prediction of treatment response in patients with autoimmune epilepsy:review of the literature and suggestions for clinical management[J].Epilepsia,2013,54(Suppl 2):48-55.
[15]Sinclair AJ,Wienholt L,Tantsis E,et al.Clinical association of intrathecal and mirrored oligoclonal bands in paediatric neurology[J].Dev Med Child Neurol,2013,55(1):71-75.
[16]Dale RC,Brilot FFagan E,et al.Cerebrospinal fluid neopterin in paediatric neurology:a marker of active central nervous system inflammation[J].Dev Med Child Neurol,2009,51(4):317-323.
[17]Baumgartner A,Rauer S,Mader I,et al.Cerebral FDG-PET and MRI findings in autoimmune limbic encephalitis:correlation with autoantibody types[J].J Neurol,2013,260(11):2744-2753.
[18]Dubey D,Samudra N,Gupta P,et al.Retrospective case series of the clinical features,management and outcomes of patients with autoimmune epilepsy[J].Seizure,2015,29:143-147.
[19]Duchowny M.Targeting tubers in paediatric epilepsy surgery candidates[J].Brain,2016,139(10):2583-2586.
[20]McKeon A.Immunotherapeutics for autoimmune encephalopathies and dementias[J].Curr Treat Options Neurol,2013,15(6):723-737.
1003-2754(2017)06-0574-03
R742.1
2017-04-12;
2017-06-03
(吉林大学白求恩第一医院神经内科和神经科学中心,吉林 长春 130021)
林卫红,E-mail:linweihong321@126.com
猜你喜欢
中国肿瘤临床(2022年4期)2022-12-12
中国现代医生(2022年19期)2022-11-04
现代临床医学(2022年1期)2022-02-12
昆明医科大学学报(2021年4期)2021-07-23
天津医科大学学报(2021年2期)2021-03-29
中华养生保健(2020年9期)2021-01-18
中国现代神经疾病杂志(2021年6期)2021-01-02
临床肝胆病杂志(2020年2期)2020-12-14
中国生殖健康(2020年7期)2020-12-10
家庭科学·新健康(2019年1期)2019-03-06
