
组织细胞坏死性淋巴结炎1例诊治分析
2017-07-18 11:19:52李春年吴小军
临床肺科杂志 2017年8期
李春年 吴小军
组织细胞坏死性淋巴结炎1例诊治分析
李春年 吴小军
组织细胞坏死性淋巴结炎(Histiocytic necrotizing lymphadenitis,HNL)是一种非肿瘤性的以淋巴结肿大为主要临床表现的自限性疾病,因其临床表现无特异,发病率低,容易导致误诊,抗生素治疗无效,对糖皮质激素敏感,预后良好。以下为1例我们在临床中误诊的病例,现报道如下。
病例资料
患者,女,49岁,农民。因“皮疹、发热18天”于2016年7月6日入住我院。患者18天前无明显诱因出现皮疹、发热,为全身红色斑丘疹,伴瘙痒。发热多于下午发生,最高体温达40度以上,伴有畏寒、寒颤、头痛、颈部疼痛,无咳嗽、咳痰、胸闷、心悸、腹痛、关节痛等。于外院抗感染治疗(头孢呋辛、替考拉宁)后皮疹较前明显消退,但仍有发热,遂就诊于我院。入院查体:BP 102/68mmHg、R 20次/分、P 80次/分、T 36.7℃,神清,背部可见散在红色斑丘疹,双侧颈部可触及数个肿大的淋巴结,最大约为3×3cm,双肺呼吸音稍粗,未闻及干湿啰音,心律齐,未及杂音,腹软,无压痛及反跳痛,肝脾肋下未及。
辅助检查:6月30日外院颅脑+胸部+腹部+盆腔CT示双侧腋下淋巴结增多,部分增大,余未见明显异常;浅表淋巴结超声示双侧颈部、腋窝、腹股沟区淋巴结肿大。7月6日外院血培养示凝固酶阴性葡萄球菌;7月8日外院血培养示人葡萄球菌人亚种。入院血常规WBC 5.41×109/L,RBC 3.35×1012/L,HB 93g/L,PLT 192×109/L,CRP 15.30mg/L;PCT 0.406ng/mL;CMV-IgG(+),EB病毒抗体NA-IgG(+),EB病毒抗体VCA-IgG阳性(+);甘油三酯及总胆固醇升高;Fe 3.5umol/L,FER 627.70ng/mL;肝肾功能、凝血、风湿三项、抗核抗体谱、4次血培养、骨髓检查均阴性。7月13日浅表淋巴结彩超示双侧颈部、腋下、腹股沟淋巴结多发肿大,其中右颈部最大淋巴结约3.4×1.0cm。7月18日ALT 124U/L,AST 200U/L。7月19日我院淋巴结活检病理结果示非霍奇金淋巴瘤,结合免疫组化考虑为外周T细胞淋巴瘤,非特殊型(PTCL-NOS)。切片送至同济医院及湖北省肿瘤医院病理科病理结果示右颈部淋巴结T区反应性增生。7月20日血常规PLT 26×109个/L;7月25日FIB 0.83g/L;血常规HB 59g/L。7月28日胸部CT示两肺多发感染。8月16日复查胸部CT恢复正常;颈部淋巴结彩超示双侧颈部淋巴结较前缩小。
入院后给予抗感染治疗,患者仍有持续性发热,波动于39-41℃,每次予以非甾体类药物退热后数小时体温再次升高,背部、双下肢、颜面部皮疹较前增多。淋巴结活检提示外周T细胞淋巴瘤非特殊型(PTCL-NOS),病理科主任会诊考虑诊断淋巴瘤依据不足,遂将切片送至外院会诊后考虑(右颈部)T区反应性增生。全院会诊后加用地塞米松5mg/d治疗,患者体温逐渐恢复正常,皮疹逐渐消退。数日后再次出现发热伴皮疹,遂将地塞米松加量为10mg/d,体温恢复正常,皮疹消退。8月16日北京友谊医院病理科会诊意见考虑(右颈部)淋巴结HNL院外随访2个月患者未再发热,结合患者临床表现及治疗反应明确诊断为组织细胞坏死性淋巴结炎。
讨 论
组织细胞坏死性淋巴结炎(HNL)是一种非肿瘤性的以淋巴结肿大为主要临床表现的自限性疾病。临床发病率低,好发于日本40岁以下年轻女性,男女发病率约为1:2[1]。HNL发病机制尚不明确,最新的研究显示HNL的发病机制可能与病毒感染机体后刺激T淋巴细胞,引起机体过度免疫反应有关[2]。
该病临床表现缺乏特异性,常急性或亚急性起病,多以痛性浅表淋巴结肿大为首发症状,以颈部淋巴结肿大最常见,大小常波动于0.5cm-4cm[3、5]。部分患者可表现为长期低热,伴或不伴上呼吸道感染症状,部分病人也因此就诊于呼吸内科[3];少数患者可伴发非特异性皮疹。实验室检查常可见白细胞正常或减少,血沉及C反应蛋白升高,可出现肝功能、乳酸脱氢酶、心肌酶异常,部分患者可合并一过性噬血细胞综合征[6]。最终诊断依靠淋巴结活检,典型光镜下病变主要位于淋巴结副皮质区,可见大片凝固性坏死,坏死边缘带可见大量的组织细胞增生、吞噬碎片、核碎裂,有些可出现新月体样组织细胞,病变中无中性粒细胞和嗜酸性粒细胞聚集,缺乏浆细胞等[1,4-5]。根据疾病的进展可将其分为增生型、坏死型和黄色瘤样型。免疫组化CD68、CD4、MPO和CD123 常阳性[1]。
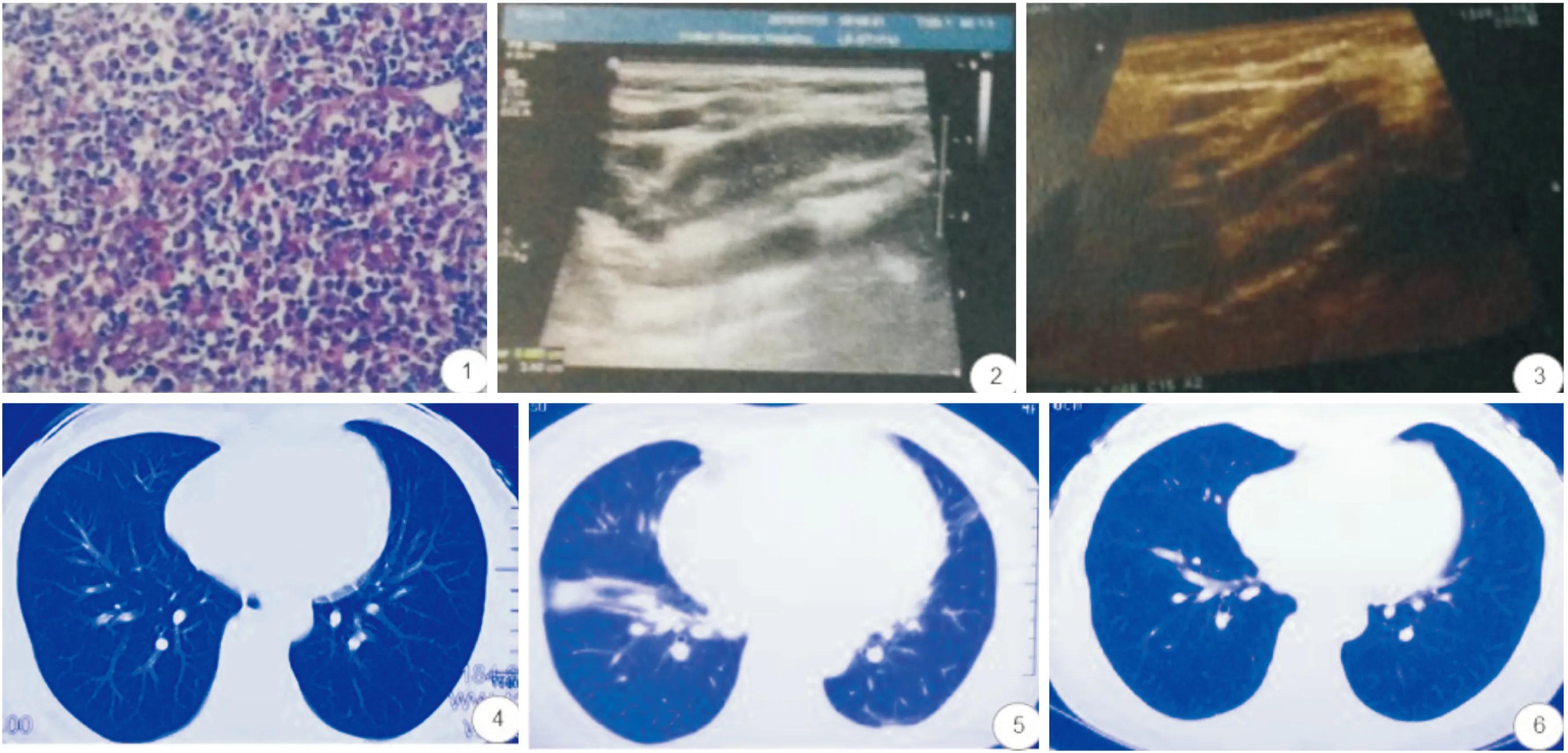
图1 镜下所见:(右颈部)淋巴结结构破坏,可见少许模糊的结节,被膜下可见灶状不规则的淡染区,伴坏死,大量胞浆丰富组织细胞及吞噬大量核碎片,周围可见不等量的小淋巴细胞、浆细胞及活化淋巴细胞;免疫组化:CD2(+),CD3(+),CD5(+),CD4(+),CD8(+),CD20灶性(+),Pax-5(-),CD68(+),CD10(-),Bcl-6(-),CD21(FDC网+),Bcl-2(+),CD30(部分+),Cyclin-D1(-),GranzymeB(+),TIA(+),CD43(+),CD56(-),CXCL-13(-),ALK(+),MPO(+),CD123(+),CD163(+),Ki67LI约30%,EBER原位杂交(-) HE染色 高倍放大 图2 7月13日颈部淋巴结彩超示可见数个椭圆形和类圆形淋巴结回声,其中较大一个大小约3.4×1.0cm 图3 8月16日颈部淋巴结彩超示可见数个椭圆形淋巴结回声,其中较大一个大小约1.3×0.4cm 图4 6月30日胸部CT示未见异常 图5 7月28日胸部CT示两肺多发感染 图6 8月6日胸部CT示未见异常
HNL为自限性疾病,自然病程约为1-6个月,大多数患者不需要特殊治疗,可自行缓解。症状严重、合并淋巴结外病变或多系统损害的患者常需药物治疗,但常规抗感染及抗结核治疗无效,糖皮质激素及羟氯喹治疗有效,预后良好,但仍有部分病例可复发,且已有死于该疾病的报道[7]。少数患者后期可发展为系统性红斑狼疮。
该患者两次外院血培养阳性,为凝固酶阴性葡萄球菌不同菌属。结合两次血培养为不同细菌,且凝固酶阴性葡萄球菌属为皮肤常驻菌,两次药敏试验提示抗生素均敏感,但是使用敏感抗生素治疗后临床表现未见好转,故考虑其血培养结果存在假阳性可能。患者院外胸部CT未见异常,住院期间复查胸部CT提示两肺感染,继续抗感染治疗后复查胸部CT 未见异常,考虑为医院获得性肺炎,也是导致误诊的另一因素。目前研究认为细菌感染、肺部感染与该病的发病关系不大,且患者在院检查提示CMV-IgG及EBV-IgG阳性,提示患者既往巨细胞病毒、EB病毒感染,符合目前HNL可能与病毒感染有关的推测。
患者首次病理结果误诊为PTCL-NOS。该病是一种高度恶性淋巴造血系统肿瘤,中老年男性常见,常表现为淋巴结进行性肿大,就诊时常伴淋巴结外侵犯及全身症状,诊断主要依靠病理检查,为排除性诊断[9]。其病理形态多样,主要表现为淋巴结结构破坏,瘤细胞形态多样、大小不一且弥散分布,细胞核形态多样不规则,偶可见RS细胞,常伴炎性背景。免疫组化常表现为CD2、CD3、CD4、CD8阳性,部分CD43、CD45RO阳性。HNL在增生期和坏死期,因出现大量增生组织细胞、浆细胞样单核细胞和免疫母细胞及核碎片,容易误诊为淋巴瘤,尤其是外周T细胞淋巴瘤。但该患者于治疗期间复查浅表淋巴结提示较前明显缩小,不符合PTCL-NOS的特征。因此在临床工作中如果病理结果与临床表现不符,应再次请病理科会诊或多次行淋巴结活检,避免造成误诊,临床上已有将HNL误诊为非霍奇金淋巴瘤并进行治疗的文献报道。
患者于治疗期间持续性高热,出现血小板、血红蛋白下降及低纤维蛋白原血症、肝功能异常、三酰甘油升高、铁蛋白升高,考虑存在继发性噬血细胞综合征[8]。然而患者于治疗期间行骨髓穿刺未见明显异常,考虑是由于骨髓穿刺时间位于血小板、纤维蛋白原下降之前,而噬血细胞综合征是一个动态过程,早期检查骨髓象可能仅存在少量噬血细胞,容易存在漏诊。
HNL的临床表现缺乏特异性,容易导致误诊,因此临床工作中遇到以下情况时应怀疑该病:①年轻女性痛性浅表淋巴结肿大;②不明原因的持续性发热,抗感染及抗结核治疗无效;③ 不明原因白细胞减少。对怀疑有此病的病人应尽快行淋巴结活检,但避免在行淋巴结活检之前滥用糖皮质激素。
[1] Deaver D,Horna P,Cualing H,et al.Pathogenesis, diagnosis, and management of Kikuchi-Fujimoto disease[J].Cancer Control,2014,21(4):313-321.
[2] Vivekanandarajah A,Krishnarasa B,Hurford M,et al.Kikuchi's Disease: A Rare Cause of Fever and Lymphadenopathy[J].Clin Med Insights Pathol,2012,5(5):7-10.
[3] Bosch X,Guilabert A,Miquel R,et al.Enigmatic Kikuchi-Fujimoto Disease A Comprehensive Review[J].Am J Clin Pathol,2004,122(1):141-152.
[4] Bosch X,Guilabert A.Kikuchi-Fujimoto disease[J].Orphanet J Rare Dis,2006,1:18.
[5] Hutchinson CB,Wang E.Kikuchi-Fujimoto disease[J].Arch Pathol Lab Med,2010,134(2):289-293.
[6] Dumas G,Prendki V,Haroche J,et al.Kikuchi-fujimoto disease: retrospective study of 91 cases and review of the literature[J].Medicine (Baltimore),2014,93(24):372-382.
[7] Uslu E,Gurbuz S,Erden A,et al.Disseminated intravascular coagulopathy caused by Kikuchi-Fujimoto disease resulting in death: first case report in Turkey[J].Int Med Case Rep J,2014,7:19-22.
[8] Henter JI,Horne A,Aricó M,et al.HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J].Pediatr Blood Cancer,2007,48(2):124-131.
[9] Horwitz SM.Management of peripheral T-cell non-Hodgkin's lymphoma[J].Curr Opin Oncol,2007,19(5):438-443.
10.3969/j.issn.1009-6663.2017.08.051
430000 湖北 武汉,武汉大学人民医院呼吸内科
吴小军,E-mail:wuxiaojunrmyy@126.com
2016-12-08]
猜你喜欢
基层中医药(2020年1期)2020-07-27 02:44:08
基层中医药(2020年1期)2020-02-12 18:25:39
基层中医药(2018年7期)2018-12-06 09:25:46
中成药(2017年5期)2017-06-13 13:01:12
现代检验医学杂志(2015年6期)2015-02-06 01:44:03
散文百家(2014年2期)2014-08-11 15:04:05
散文百家·下旬刊(2014年2期)2014-04-10 16:24:05
四川生理科学杂志(2014年3期)2014-02-28 14:09:37
中国中西医结合外科杂志(2013年3期)2013-03-11 20:05:00
实用老年医学(2013年7期)2013-03-11 18:39:07
