
基于色谱-质谱平台的代谢组学数据预处理方法*
2017-07-18 11:08哈尔滨医科大学卫生统计学教研室150081
中国卫生统计 2017年3期
哈尔滨医科大学卫生统计学教研室(150081)
孙 琳 张秋菊 王文佶 曲思杨 谢 彪 高 兵 刘美娜△
·方法介绍·
基于色谱-质谱平台的代谢组学数据预处理方法*
哈尔滨医科大学卫生统计学教研室(150081)
孙 琳 张秋菊 王文佶 曲思杨 谢 彪 高 兵 刘美娜△
代谢组学的概念自20世纪90年代被正式提出[1],已被广泛应用于医学研究领域,其一般研究流程包括样本采集、样本检测、数据预处理、数据分析和生物学解释等。常用的样本检测技术有核磁共振(nuclear magnetic resonance,NMR)和高分辨率色谱-质谱联用技术[2],本文所述方法针对后者。经色谱-质谱联用平台检测的数据具有以下特点:高维度,小样本,变量间高度相关,高噪声,高缺失,以及高度变异性。基于以上数据特征,在代谢组学数据分析之前需要对数据进行预处理[3],以消除或减小数据中高噪声、高缺失和高度变异性对统计分析结果的干扰。数据预处理是数据分析的前提,有利于统计分析过程的多变量模型构建;若数据未经过充分预处理而直接用于统计分析,可能会掩盖数据中某些真实特征(如组间差异),使研究结果模糊化。数据预处理包括缺失数据处理,数据标准化,以及数据的中心化、标度化和转换等内容。本文将全面介绍基于色谱-质谱联用平台的代谢组学数据预处理方法,并进行各方法比较,为研究者选择合适的数据预处理方法提供思路。
数据预处理的重要性
代谢组学研究经常涉及处于不同实验条件下两组样本的分类判别(例如处理组和非处理组、病例组和对照组),目的是建立某种疾病或危险因素的预测模型,找到相关的差异表达生物标志物,辅助疾病诊断、预防或治疗[4]。代谢组学数据中存在多种变异,其中,由于某种特定诱导因素(实验干预、疾病等)导致的代谢物浓度差异被称为诱导变异,这是研究者感兴趣的变异,也是数据分析所针对的变异;此外,实验过程中其他因素引起的变异也会影响检测的代谢物峰强度,从而影响研究对象分类,主要包括非诱导变异和技术平台变异。
非诱导变异[5]包括:(1)量级差异:指机体内某个单分子物质的平均浓度远小于高浓度化合物(例如ATP)的平均浓度,从生物学角度来说,相对于低浓度的代谢物,高浓度代谢物在机体生理病理过程中未必发挥更重要的作用;(2)倍数差异:指诱导因素对不同通路的代谢物的影响程度不一致,一般来说,中心代谢途径物质的浓度较为稳定,而次级代谢途径产生的物质的浓度易受环境因素影响;(3)生物固有差异:指在相同的实验条件下,某些代谢物的浓度在同组别的个体间也展现出较大幅度波动,这种个体差异可能会导致错误的判别结果。技术平台变异来源于样本预处理过程和色谱-质谱检测平台,例如预处理时样本间细微的体积差异、质谱离子源的波动和色谱柱效能的变化等。检测平台变异导致测量误差,具体表现为数据中的噪声,在统计分析时,通常假定代谢物噪声强度呈正态分布,在实际研究中这种假设往往不成立,偏态分布的噪声造成数据的异方差性,影响统计分析结果。
数据预处理的目的就是减少诸多不利因素对多变量模型建立和生物标志物筛选的干扰,以保证研究结果的准确性。代谢特征数据由色谱-质谱平台输出后,要经过原始数据的预加工和清洁数据的预处理,才能进入数据分析阶段。
清洁数据
在数据预处理前,首先需要将原始数据转换为清洁数据。生物样本经色谱-质谱联用平台检测得到由质荷比、保留时间和代谢物相对峰强度信息构成的三维原始数据,这种数据杂乱且不匹配,不能直接反映研究对象的代谢特征。首先要对原始数据进行预加工,包括保留时间对齐、峰提取和峰匹配等步骤,把原始数据转换成匹配的色谱-质谱-代谢物数据。预加工后的数据矩阵由几十到上百个样本(观测)所对应的上千个代谢物(变量)的质谱峰强度构成,称为清洁数据。实际研究中,预处理和统计分析阶段的原始数据指的是清洁数据而非仪器导出的原始数据。图1展示了整个数据预处理的流程。
常用的代谢组学数据预处理方法
1.缺失值处理 色谱-质谱联用平台输出数据中存在大量“0”值,我们把这些“0”值标记为缺失数据。产生缺失值的原因有三方面[6]:代谢物只存在于某些样本,在其他样本中浓度为0;代谢物存在于样本中,但其浓度低于检测仪器设定的检出限;代谢物在样本中的浓度高于检出限而被色谱检出,但没有得到正确的质谱峰匹配,在质谱中无法检出。缺失值的产生会对深入数据挖掘结果产生较大偏倚,降低统计分析的效率,因此需要对缺失数据进行删减和填补。
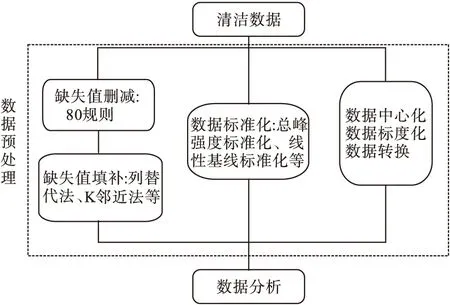
图1 代谢组学数据预处理流程图
(1)缺失值删减 一般遵守“80规则”[6],即:如果某一代谢物在一个类别超过80%的样本中的检测强度不为0,则该变量予以保留;反之剔除。通过“80规则”,由于检测仪器导致的数据缺失被移除。
(2)缺失值填补 在初步缺失值删减后,数据中仍存在一部分“0”值,需要对这部分数据进行填补。目前常用的缺失值填补方法包括列替代法[7-8]、K邻近法[8-9]、贝叶斯主成分分析法[7]和多重填补法[10-12]等。
列替代法 由于研究对象的同质性,代谢物在同类别研究对象的不同个体间的代谢行为高度相似。因此,某种代谢物在特定实验条件下的缺失值,可用缺失数据所在列的其他样本的数据估计缺失的代谢物强度,包括最小值替代法、均值替代法和中位数替代法。列替代法简单易操作,但是并没有考虑到数据中各变量的相关性,因此估计的准确度较低。
K邻近法 首先计算含有缺失值的代谢物和所有其他代谢物间的欧氏距离,选取与缺失变量欧氏距离最近的K个变量的加权平均峰强度值填补缺失数据, K选择10至20较为合理。不同于列替代法,K邻近填补法对每个缺失值给出不同估计值,更接近真实数据,而且考虑了变量间的相关性。
贝叶斯主成分分析法(Bayesian principal components analysis,BPCA) 此种填补方法是基于主成分回归、贝叶斯估计和EM算法的三层算法。先用主成分分析的结果建立完整数据的主成分回归模型,用回归模型的预测值作为缺失数据的初步估计值,建立贝叶斯估计的先验分布,假定残差和代谢物在主成分上的投影都是正态独立的变量,分布参数未知,由EM算法迭代估计上述参数直至收敛,用最终的主成分回归模型预测值作为缺失数据的估计值。BPCA填补的优势在于利用了数据的全部结构,对样本量较大的随机缺失处理效果较好[28-29]。
多重填补法 与单一填补不同,多重填补用某个缺失值的可能取值的集合进行填补,重复多次,产生多个完整数据集;随后对多个数据集进行统计分析,得到每个缺失数据的平均水平和变异水平;最后,整合来自各填补数据集的结果,从而得到统计推断的结果。该方法考虑到了缺失数据的不确定性对统计推断结果的影响。
以上介绍的方法侧重点不同,实际研究中,判断是否需要进行缺失值填补以及选择何种填补方法需要研究者依据数据特征决定。
2.数据标准化(表1) 标准化针对代谢组学实验中的系统误差[13],包括样本采集过程中的差异(如尿液样本采集时间不同)、个体差异(如由于饮水量不同而导致的尿液浓度差异)或大规模研究中的批次差异。与其他数据预处理方法相比,标准化方法更为复杂 ,这里简述几种常用的标准化方法。
(1)总强度标准化 假定数据中所有样本的全部特征的峰强度总和相等,即所有代谢物的总强度在样本之间不发生变化。每个观测中所有元素的标准化因子相同,将原始数据除以所有特征的峰强度之和得到新数据[14]。这是代谢组学中最常用的标准化方法,Waters公司的软件MakerLynx中默认标准化方法就是总强度标准化。
(2)Cyclic Loess标准化 其基本思想是通过曲线拟合的方法,调整两个样本间的所有特征的对数峰强度值差值,使之近似为0[15-16]。Cyclic Loess对不同类别样本数据分别进行标准化,可以有效地减少组间个体变异和技术平台变异。
(3)Quantile标准化 是基因组学中常用的标准化方法[18],目的是使全部样本的数据具有相同分布[17],当利用Q-Q图对数据进行可视化时,所有点近似分布于对角线上。通过使各样本数据分位数相等,达到所有观测代谢物数据的一致分布。
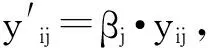
3.数据的中心化、标度化和转换(表2-3)
对于非诱导变异和技术变异,除上述标准化方法外,还可以采取中心化和标度化处理。转换则是对由于噪声导致的数据异方差性进行处理。通过这一阶段的预处理,数据中来源于诱导因素的代谢物浓度变异得以突显。

表1 标准化方法总结
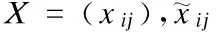
(1)数据中心化
具体方法是将原数据减去每个列变量的均值而得到新数据,中心化后,新数据围绕0上下波动,而不再围绕代谢物峰强度均值波动。通过中心化,高浓度代谢物和低浓度代谢物间的浓度差异得到适当的调整,突出数据中波动部分的重要性[19-20]。中心化是标度化和转换的基础,以下提到的两类方法都要与中心化相结合。
(2)数据标度化
Auto scaling 自标度化,又称为单位标度化或单位方差标度化。以标准差作为标度因子,用中心化后的数据除以列变量的标准差而得到新数据[20]。自标度化后,所有代谢物强度的标准差均为1,相当于把所有的变量置于同等重要的水平,消除了由于代谢物的绝对含量差异引起的高浓度物质对低浓度物质的数据掩盖。需要注意的是,自动标度化给所有代谢物以相同权重,使低含量代谢物更可比,但是,这些物质的测量误差同时被放大 ,可能会导致错误的数据分析结果。
Range scaling 极差标度化,采用生物学范围作为标度因子[21],这里的生物学范围指的是某种代谢物强度的最大值与最小值之差,即列变量的极差。标准差涵盖了所有研究对象的信息,而极差的计算只用到两个样本的信息, 因此极差标度化对于数据中的离群值很敏感。为了提高方法的鲁棒性,可以采用其他更稳健的生物学范围计算方法。
Pareto scaling 该方法和自动标度化类似,只是标度因子由标准差变为标准差的平方根[22]。Pareto标度化在弱化高含量代谢物重要性的同时又保持了数据的完整性,与自动标度化方法相比,新数据更接近原始测量值。与在组别间变化不显著的代谢物相比,变异较大的物质对此方法更加敏感。
Vast scaling 大规模标度化,又称为变量稳定性标度化,是自标度化的扩展[23]。大规模标度化重点关注那些在样本间变异较小,浓度较稳定的代谢物,采用标准差和变异系数作为标度因子。变异系数是列变量标准差和均数的比值,引入变异系数作为标度因子,提高了低相对标准偏差变量的重要性,降低了高相对标准偏差变量的重要性。大规模标度化既可用于无监督模式识别,也可用于有监督模式识别,当进行有监督模式识别时,用各组的信息分别求组内变异系数作为标度因子。
Level scaling 水平标度化,使用的标度因子是代谢物的平均强度[5],标度化后的新数据是某种代谢物强度相对于其平均强度变化的百分比,即相对变化值。水平标度化适用于研究特定的某些相对变化大的生物学反应(如应激反应)和高浓度生物标志物的发现。
(3)数据转换
转换是非线性的数据预处理方法,主要用于校正数据中的噪声结构[24],通过数据转换,将乘性噪声转换为加性噪声,使偏态分布的数据转换为更加对称的分布。
经UPLC-MS检测输出的峰强度包括代谢物的信号强度以及噪声强度这两部分,其中噪声具有乘性结构或加性结构[25-26],可以用以下模型表示:
xij=βij+nij·sijeηij
xij是检测到的第i种代谢物在第j个样本中的峰强度,sij是预期的峰强度,βij表示随机背景噪声(电子噪声),ηij表示乘性随机噪声(例如样本预处理过程出现的变异,离子源的波动或样品导入设备的波动),ηij表示样本j的标准化因子。加性噪声是基线的随机波动,与代谢物的信号强度无关,来自于检测仪器的电子噪声,由于交叉进样,加性噪声均匀出现在各组别的全部样本的检测过程中,因此可忽略不计。与此相反,乘性噪声随代谢物信号强度增加而增强,通常噪声强度与信号强度成比例。由于乘性噪声的存在,进行不同样本的多次测量时,高浓度的代谢物会展现出更大的变异,造成某些低浓度代谢物的信号可能会被高浓度代谢物的强噪声所掩盖。

表2 数据中心化和数据标度化方法的总结
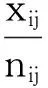
对数转换 是代谢组学常用的数据转换方法。如果数据中各变量的相对标准偏差是一个常量,则使用对数转换能完全消除乘性噪声对代谢物峰强度的累加作用[24],在实际研究中,这种情况非常罕见。对数转换无法处理数据中的零值,因此对数转换前先要对数据进行缺失值填补。对数转换的另一个缺陷是不能很好地处理那些峰强度相对标准偏差很大的代谢物,这些物质通常有较低的相对浓度,变异相对于均数更加突出,当峰强度趋于0时,对数转换后的数值接近负无穷。
平方根转换 对于缺失值较多或低浓度代谢物较多的数据,通常选择平方根转换[27]来校正噪声强度。平方根转换后,高浓度代谢物的方差明显减小,使得浓度不同的代谢物的方差近似相等。平方根转换只是缩小了乘性噪声强度,并没有将其转变为加性噪声。

表3 数据转换方法总结
小 结
在代谢组学研究过程中,数据预处理会对统计分析结果产生很大影响,因此数据分析前的预处理过程是必不可少的。预处理方式多样,关于预处理方法的比较研究较少 。本文介绍了多种数据缺失值填补法,标准化法以及标度化、转换法。
从理论上看,BPCA填补法和多重填补法对缺失值的估计利用了数据集中全部的信息,填补效果更好。Hrydziuszko等[30]比较各种缺失值填补方法(包括基于单变量方法和基于多变量的方法)处理代谢组学数据的效果,发现K邻近法的填补效果最优。
Bedilu[31]等比较不同标准化方法对于大规模代谢组学数据的影响,发现曲线拟合的Cyclic Loess标准化法能最有效地移除系统误差。Kévin[32]等通过对实验数据的分析发现,在几种标准化方法中,Cyclic Loess效果较好,线性的标准化法效果较差。Pirtr S[33]等比较不同标度化方法基于主成分-判别分析、随机森林和K最邻近分类三种多变量分析方法对于统计分析结果的影响,发现自动标度化和极差标度化优于其他方法,Robert等[5]也得到了类似的结论。
数据转换对大数值的影响相较于小数值更大,缩小了高浓度代谢物和低浓度代谢物浓度之间的量级差异,起到了类似标度化的伪标度化作用。大规模大样本量的代谢组学研究涉及到多批次数据的合并,数据中的噪声结构更为复杂,可以考虑标度化和转换两种方法的结合运用。在高维组学数据中,数据转换和标度化之间的相互影响很复杂,应用何种方法还要依据实际情况而定。
预处理方法选择不仅取决于代谢数据的特点,还与之后选用的数据分析方法有关。例如,聚类方法关注于特征间相似点(或不同点)的分析,而主成分分析则是试图用尽可能少的成分解释数据中大部分变异(降维);数据预处理可能会改善聚类方法的结果,却使主成分分析的结果模糊化 。值得注意的是,上述预处理方法比较研究的结论都基于研究者在特定条件下模拟得到的数据集和特定的实验数据集的分析而得出的,在外推到其他数据集时要慎重。选择数据预处理方法时要综合考虑研究目的、数据结构特征和拟采用的统计分析方法,再决定预处理的策略和具体方法。
[1]Oliver SG,Winson MK,Kell DB,et al.Systematic functional analysis of the yeast genome.Trends in biotechnology,1998,16(9):373-378.
[2]Salek RM,Steinbeck C,Viant MR,et al.The role of reporting standards for metabolite annotation and identification in metabolomic studies.GigaScience,2013,2(1):1.
[3]Goodacre R,Broadhurst D,Smilde AK,et al.Proposed minimum reporting standards for data analysis in metabolomics.Metabolomics,2007,3(3):231-241.
[4]Fiehn O.Metabolomics-the link between genotypes and phenotypes.Plant molecular biology,2002,48(1-2):155-171.
[5]van den Berg RA,Hoefsloot HCJ,Westerhuis JA,et al.Centering,scaling,and transformations:improving the biological information content of metabolomics data.BMC genomics,2006,7(1):1.
[6]Bijlsma S,Bobeldijk I,Verheij ER,et al.Large-scale human metabolomics studies:a strategy for data(pre-)processing and validation.Analytical chemistry,2006,78(2):567-574.
[7]Xia J,Psychogios N,Young N,et al.MetaboAnalyst:a web server for metabolomic data analysis and interpretation.Nucleic acids research,2009,37(suppl 2):W652-W660.
[8]Steuer R,Morgenthal K,Weckwerth W,et al.A gentle guide to the analysis of metabolomic data.Metabolomics:Methods and protocols,2007:105-126.
[9]Troyanskaya O,Cantor M,Sherlock G,et al.Missing value estimation methods for DNA microarrays.Bioinformatics,2001,17(6):520-525.
[10]Rubin DB.Multiple imputations in sample surveys-a phenomenological Bayesian approach to nonresponse//Proceedings of the survey research methods section of the American Statistical Association.American Statistical Association,1978,1:20-34.
[11]Schafer JL.Multiple imputation:a primer.Statistical methods in medical research,1999,8(1):3-15.
[12]Buuren S,Groothuis-Oudshoorn K.mice:Multivariate imputation by chained equations in R.Journal of statistical software,2011,45(3):.
[13]Kultima K,Nilsson A,Scholz B,et al.Development and evaluation of normalization methods for label-free relative quantification of endogenous peptides.Molecular & Cellular Proteomics,2009,8(10):2285-2295.
[14]Veselkov KA,Vingara LK,Masson P,et al.Optimized preprocessing of ultra-performance liquid chromatography/mass spectrometry urinary metabolic profiles for improved information recovery.Analytical chemistry,2011,83(15):5864-5872.
[15]Cleveland WS,Devlin SJ.Locally weighted regression:an approach to regression analysis by local fitting.Journal of the American Statistical Association,1988,83(403):596-610.
[16]Dudoit S,Yang YH,Callow MJ,et al.Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments.Statistica sinica,2002:111-139.
[17]Bolstad BM,Irizarry RA,Åstrand M,et al.A comparison of normalization methods for high density oligonucleotide array data based on variance and bias.Bioinformatics,2003,19(2):185-193.
[18]Hansen KD,Irizarry RA,Zhijin WU.Removing technical variability in RNA-seq data using conditional quantile normalization.Biostatistics,2012,13(2):204-216.
[19]Bro R,Smilde AK.Centering and scaling in component analysis.Journal of Chemometrics,2003,17(1):16-33.
[20]Jackson JE.A user's guide to principal components.1991.
[21]Smilde AK,van der Werf MJ,Bijlsma S,et al.Fusion of mass spectrometry-based metabolomics data.Analytical chemistry,2005,77(20):6729-6736.
[22]Eriksson L.Introduction to multi-and megavariate data analysis using projection methods(PCA & PLS).Umetrics AB,1999.
[23]Keun HC,Ebbels TMD,Antti H,et al.Improved analysis of multivariate data by variable stability scaling:application to NMR-based metabolic profiling.Analytica chimica acta,2003,490(1):265-276.
[24]Kvalheim OM,Brakstad F,Liang Y.Preprocessing of analytical profiles in the presence of homoscedastic or heteroscedastic noise.Analytical Chemistry,1994,66(1):43-51.
[25]Anderle M,Roy S,Lin H,et al.Quantifying reproducibility for differential proteomics:noise analysis for protein liquid chromatography-mass spectrometry of human serum.Bioinformatics,2004,20(18):3575-3582.
[26]Durbin BP,Hardin JS,Hawkins DM,et al.A variance-stabilizing transformation for gene-expression microarray data.Bioinformatics,2002,18(suppl 1):S105-S110.
[27]Sokal RR,Rohlf FJ.Assumptions of analysis of variance.Biometry:The principles and practice of statistics in biological research,1995:392-450.
[28]Albrecht D,Kniemeyer O,Brakhage AA,et al.Missing values in gel-based proteomics.Proteomics,2010,10(6):1202-1211.
[29]Oba S,Sato M,Takemasa I,et al.A Bayesian missing value estimation method for gene expression profile data.Bioinformatics,2003,19(16):2088-2096.
[30]Hrydziuszko O,Viant MR.Missing values in mass spectrometry based metabolomics:an undervalued step in the data processing pipeline.Metabolomics,2012,8(1):161-174.
[31]Ejigu BA,Valkenborg D,Baggerman G,et al.Evaluation of normalization methods to pave the way towards large-scale LC-MS-based metabolomics profiling experiments.Omics:a journal of integrative biology,2013,17(9):473-485.
[32]Contrepois K,Jiang L,Snyder M.Optimized analytical procedures for the untargeted metabolomic profiling of human urine and plasma by combining hydrophilic interaction(HILIC)and reverse-phase liquid chromatography(RPLC)-Mass spectrometry.Molecular & Cellular Proteomics,2015,14(6):1684-1695.
[33]Gromski PS,Xu Y,Hollywood KA,et al.The influence of scaling metabolomics data on model classification accuracy.Metabolomics,2015,11(3):684-695.
(责任编辑:邓 妍)
国家科技支撑计划(2011BAI09B02);黑龙江省自然基金重点项目(ZD201314)
△通信作者:刘美娜,E-mail: liumeina369@163.com
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中山大学学报(自然科学版)(中英文)(2022年4期)2022-08-05
昆明医科大学学报(2022年3期)2022-04-19
昆明医科大学学报(2021年4期)2021-07-23
智慧健康(2021年33期)2021-03-16
天津医科大学学报(2021年1期)2021-01-26
四川大学学报(自然科学版)(2020年3期)2020-06-03
中国医院用药评价与分析(2020年3期)2020-05-29
中国建材科技(2020年6期)2020-03-23
农药科学与管理(2019年5期)2019-08-13
