
磷酸二酯酶6b基因异常表达对视网膜色素变性的作用
2017-07-18 11:45袁莉,刘焰*,刘特
中国临床医学 2017年3期
袁 莉, 刘 焰*, 刘 特
1.上海交通大学医学院附属第一人民医院眼科,上海 200080 2.上海市中医老年病研究所,上海 200031
·综 述·
磷酸二酯酶6b基因异常表达对视网膜色素变性的作用
袁 莉1, 刘 焰1*, 刘 特2
1.上海交通大学医学院附属第一人民医院眼科,上海 200080 2.上海市中医老年病研究所,上海 200031
磷酸二酯酶(phosphodiesterase,PDE)是一个大家族,包括11个亚家族,即PDE1~PDE11。而PDE6是PDEs亚家族中的一种,由PDE6基因编码的PDE蛋白,水解cGMP,cGMP是视杆细胞外节膜盘离子通道的特异性受体,同时cGMP也是脊椎动物中感光细胞将光信号转化为电信号的重要分子。PDE6b基因的缺失使感光细胞内cGMP增多,引起感光细胞的阳离子增多,细胞中毒而坏死,视网膜的感光细胞不断破坏,从而间接影响到视网膜接受光信号转换为电信号。PDE6b基因异常是常染色体隐性遗传视网膜色素变性最主要的原因之一,因此对于PDE6b基因表达异常在动物模型及家族遗传逐渐成为近年来研究的热点。该文将重点阐述PDE6b基因的结构、功能和突变类型对视网膜色素变性相对应结构及其功能的影响。
磷酸二酯酶β亚单位;视网膜;rd1;基因缺失;视网膜色素变性
PDE6b基因最早是在rd1小鼠中被发现,其编码的PDE6β是视网膜感光细胞将光量子转化为电信号的重要组成部分,引起视网膜的级联反应。生物信息分析显示,PDE6b基因任何位点上的突变都会影响到PDE6b三维立体结构,导致PDE蛋白结构发生改变,从而引发其功能失调。PDE6b基因表达异常被认为与常染色体显性遗传、常染色体隐性遗传视网膜色素变性以及先天性静止性夜盲症等疾病相关。随着研究的深入,人们逐步认识到PDE6b基因在视网膜光电转换中的重要作用,尤其PDE6b基因的缺失引起视网膜色素变性等疾病。本文就PDE6b基因的结构功能及其能引起视网膜色素变性的突变类型进行阐述。
1 PDE6b结构及功能
1.1 PDE6b基因的结构 Keeler等[1]于1924年首次报道了视网膜无杆状细胞的小鼠,后被作为研究常染色体隐性遗传视网膜色素变性的动物模型。Pittler等[2]将这类小鼠简称为rd1小鼠,rd1小鼠携带缺陷的PDE6b基因(ID:5158),PDE6b位于小鼠的5号染色体,rd1小鼠的产生是由PDE6b基因上一个鼠白血病病毒在内含子1位的插入和一处点突变产生终止密码子所造成的。Pittler等[3]于2001年证实,在外显子7位(密码子347)有处无义点突变,和Keeler等[1]所发现的突变类型在同一个基因的不同位点。虽然视网膜色素变性的PDE6b基因表现型最早在欧洲的野生小鼠中被观察到,但后来也被发现存在于多种野生及实验室近交鼠系中。与人类同源的PDE6b基因与人类的常染色体隐性遗传的视网膜色素变性相关。犬类PDE6b基因位于3号染色体上,约有15 kb,与人类不同的是,该基因在内含子3、10、11、12、21片段更短一些,外显子14位有110个碱基,除此之外,外显子15位上有88个碱基比人(87)稍长。
PDE6b基因定位于人类染色体4p16.3,含22个外显子,约有45 kb,cDNA有3 231个碱基,编码855个氨基酸,将该区域精确定义至2.5 Mb,PDE6b基因主要表达于感光细胞的外段,并证实PDE6b基因表达异常为视网膜色素变性(retinitis pigmentosa,RP)的致病基因之一。RP从遗传学角度来讲有多种类型的遗传方式,比如常染色显性遗传、常染体隐性遗传或者X连锁遗传。
1.2 PDE6b基因的功能 PDE6b基因编码PDE6蛋白的β亚基,PDE6包含两大亚基PDE6α(88 KDa)和-β(84 KDa),每一个都有一个催化区和两个结合区,另外还有两个起抑制作用的PDE6γ亚基藏于G蛋白空隙中。生物信息学分析,PDEβ亚单位二级结构由2个GAF(GMP phosphodiesterase/adenyl/cyclase/FhlA)结构域和一个PDEaseⅠ(phosphodiesterase Ⅰ)结构域组成。GAF结构域在视紫红质(rhodopsins,RHO)和环核苷酸磷酸二酯酶(cyclic nucleotide phosphodiesterase,cGMP-PDE)中特异性存在;PDEaseⅠ结构域是酶催化活性中心,这2种结构域上的异常可以引起PDE功能的异常。迄今已发现PDE有11个亚族,结构和功能分析表明,PDE拥有一个C端催化结构域、N端调节结构域和一个末端的高度保守的半胱氨酸富集区域(cysteine rich domain,CRD)。磷酸二酯酶在光传导过程中发挥着重要作用,外界光子的刺激使视紫红质活化分解为视黄醛和视蛋白,同时视紫红质激活光转导蛋白(transducin),活化的转导蛋白又激活了视杆细胞中PDE,cGMP-PDE降解感光细胞内环磷酸鸟苷(cGMP)转化为5′-GMP,使cGMP的浓度降低。cGMP是视杆细胞外节膜盘离子通道的特异性受体,cGMP是脊椎动物中感光细胞将光反应转化为电信号的重要分子[4],它的降解导致视杆细胞膜阳离子依赖的cGMP通道关闭,Na+、Ca2+内流减少,感光细胞质膜发生超极化,突触末梢释放谷氨酸递质增加,形成神经冲动,进而引起视觉冲动向视觉中枢逐级传递,人们因此而感受到光线的刺激(图1)。
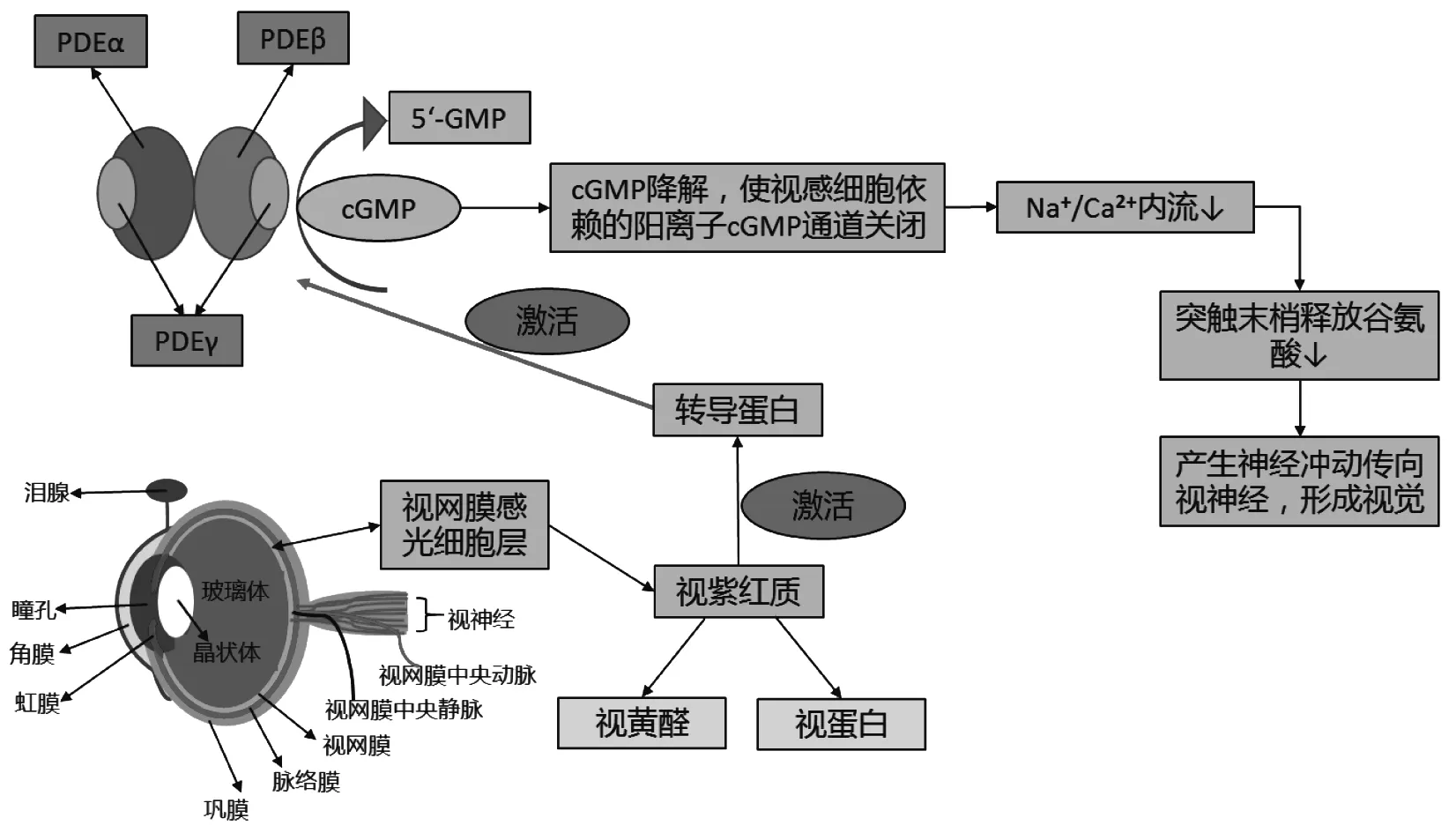
图1 PDE作用机制示意图
2 PDE6b突变与缺失对视网膜结构和功能的影响
PDE6b基因编码蛋白与视网膜光电信号转导的瀑布式反应相关,它的异常表达已知主要发生在小鼠、犬类以及人类。
2.1 PDE6b基因表达异常对动物视网膜结构和功能的影响
2.1.1 PDE6b基因表达异常对rd1小鼠和rd10小鼠的影响 Keeler等[1]发现的无视杆细胞的rd1小鼠有PDE6b基因的缺失。Pittler等[2]于1991年证实了rd1小鼠在PDE6b基因外显子7位第347个密码子处发生错义突变。rd1小鼠存在两处点突变,一处是约1.5 kb大小片段的鼠白血病病毒基因在内含子1位插入,另一处是外显子7位的第347密码子发生无义突变(C-A)产生了一个终止密码子,其中外显子7位上的无义突变是导致rd1小鼠产生的主要原因。Chang等[5]对rd1小鼠的补充实验意外地发现,在视网膜色素变性的显性病例中存在新的PDE6b等位基因,在PDE6b基因的外显子16位的第1 976位点上发生错义点突变,第659位的密码子CTC突变为CCC,亮氨酸转变为脯氨酸。这个突变的位点不同于rd1,该学者将这个等位基因的标志物称为PDE6b nmf,将其简化为nmf。Chang等[5]首次描述了rd10小鼠,rd10小鼠在PDE6b基因的外显子13位发生了错义突变。rd10小鼠为视网膜色素变性呈现出另外一个有用的自然突变的模型。除了这些自发的突变,Hart等[6]发表了一篇关于PDE6b基因缺失的基因型和表型研究,发现ENU(N-ethyl-N-nitrosourea)可以诱导PDE6b基因的7个新的位点突变,其中4个位点突变和rd1小鼠有同源性,三个无义突变(Arg799X,Trp378X和Tyr689X),一个突变改变了5’端剪接,这些使PDE蛋白的功能受到严重干扰,另外还有两个发生错义突变(H620Gln和Asn606Ser),另一个是5′端的突变(IVS+5G→A),而这种突变引起的rd1小鼠视网膜变性发展速度相较于自发的rd1小鼠比较慢。
正常哺乳动物视网膜光传导由感光细胞通过其内外节的特殊结构将光信号转化为电信号,再传给双极细胞。rd1小鼠模型中,由于鼠性病毒基因插入,编码PDEβ亚单位的PDE6b基因发生突变,编码的PDE蛋白结构发生改变,PDEase1水解cGMP的能力降低,感光细胞内cGMP的堆积,通过生物化学证实PDEβ的功能障碍和细胞内cGMP增高相关。rd1小鼠视网膜中cGMP水平升高,过高的cGMP引起细胞内钠、钙离子的过度内流,将使Na+/Ca2+-ATP酶持续活化以维持电化学梯度,从而加重了代谢负担,细胞中毒,最终造成了感光细胞的死亡。
研究[4]表明,在小鼠视网膜变性的模型中,视网膜结构和功能都受到很大的影响和破坏,并且视网膜中cGMP的水平升高的程度要高于视网膜的破坏,主要表现为眼底血管变细、大量的色素斑沉着(呈骨细胞样)、视网膜明显变薄[4]、巩膜、视网膜血管萎缩;超微结构显示,视网膜变性过程中外核层细胞核固缩,外节膜盘排列紊乱甚至崩解。病变过程中主要累及外核层的视杆细胞,晚期累及视锥细胞,视网膜色素上皮及内层视网膜细胞形态和功能保持正常。在nmf突变的纯合子小鼠身上,表现为出生后的16 d,视网膜的外核层出现大面积的破坏,30 d左右视网膜外核层所剩无几,视网膜出现白斑,视网膜血管缺血变白。rd10小鼠这种突变模型PDE6b基因编码的PDE蛋白表达水平及活性都不同于rd1小鼠,rd10小鼠视网膜的变性的发生相对于rd1小鼠发生比较晚,中心视网膜部视杆细胞的破坏出现在出生后的16 d,外周视网膜在出生后的20 d开始出现破坏,30 d左右时视网膜的外核层仅剩下两三层,所有的视杆细胞的死亡大概在60 d左右,感光细胞消失的速率要慢于rd1小鼠[7]。组织学显示,视网膜的外核状层在出生后的24 d几乎消失殆尽。这些特征性变化都与人的视网膜色素变性临床表现是相似的[8]。视网膜电图显示,视网膜电图异常,但是视杆细胞和视锥细胞的a波和b波在出生后的第18 d是很容易被测到的,在随后的两个月左右稳定的下降超过90%[8]。
2.1.2 PDE6b基因表达异常对犬类视网膜功能的影响 据报道[9],犬是PDE6b基因缺陷造成视网膜营养障碍相关的第二种动物。由于犬类与人类的基因库大体是相似的,对其进行目的基因筛查的过程中发现PDE6b基因位于3号染色体上。PDE6b基因的突变发生在犬类中最早是由Aguirre等[10]报道的。Suber等[11]发现,rcd1(rod-cone dystrophy1)狗在PDE6b基因在外显子21位第807密码子处发生无义突变,G-A密码子由TGG转变为TAG,引起终止密码子的出现,其后的49个氨基酸无法正常转录,C端的缺失造成翻译后加工及膜结合功能障碍,最终使PDE失去酶的活性。Dekomien等[12]于2000年报道的Sloughi dog,在PDE6b基因的816位密码子后有8个碱基的插入。Goldstein等[13]在American Staffordshire terrier 和the Pit Bull Terrier种系的犬类发现了两类早发性常染体隐性遗传视网膜色素变性,患有这种疾病的犬类出生后不到一年开始出现明暗视觉严重受损的情况,甚至在成年早期即发生严重的失明,这两类分别称为crd1和crd2(cone-rod dystrophy1、2),对其进行全基因组关联分析和之后的研究发现这两类疾病的致病基因并非是等位基因,crd1是PDE6b基因的缺失突变引起,crd2是因IQCB1的插入突变引起。在crd1狗中PDE6b基因的突变是在21位外显子上有3个碱基的缺失,引起最终PDE蛋白的催化区一个氨基酸的缺失,而这个氨基酸恰好位于PDEase-1酶活性区域内,PDEase-1酶催化功能区域开始于556终止于804,而这个缺失的氨基酸刚好位于802位。组织学显示,crd1狗在出生后11周左右,视网膜的外核层仅有5~7层厚,经过组织病理学染色发现视网膜内节发生很严重异性、紊乱、松弛的排列,且相对来说视杆细胞要比视锥细胞更容易发生这种变化,在20个月之后,crd1狗视网膜的感光细胞的外节、内节以及细胞核都发生很严重的损伤。生物化学分析表明,感光细胞发育受阻滞的程度与cGMP水平的异常增高相关,cGMP 水平持续增高直到正常的10倍,也可以降低cGMP磷酸二酯酶的活性。编码PDEβ的mRNA开始出现异常的低水平要早于视网膜出现破坏。在这项研究[13]中显示,两种犬类rcd1、crd1和小鼠的rd1、rd10是分别相对应的,在突变的类型、表型、发生年龄、发生率、和基因治疗的潜在性皆相似,rcd1和rd1都有一个错义突变,而crd1和rd10发生的是无义突变,并且功能学分析显示rcd1和rd1几乎有完全性的视网膜功能的破坏。
2.2 PDE6b基因表达异常对人类视网膜功能的影响 PDE6b基因的突变通常与视网膜色素变性和先天性静止性夜盲症相联系[14],也可引起感音神经性耳聋-视网膜色素变性综合征,简称为Usher综合征(Usher Syndrome,USH)[15]。McLaughlin等[16]首次报道了PDE6b基因与人类的常染色体隐性遗传的视网膜色素变性相关。视网膜色素变性是一类进行性的视网膜视杆细胞和视锥细胞的营养障碍性疾病,表现为患病者在幼儿期皆出现夜盲症、逐渐的视野缺损、视敏度下降。眼底检查示,视网膜血管旁色素沉着、黄斑区蜡白、视网膜血管进行性萎缩。视网膜电图检查显示,视杆细胞和视锥细胞波振幅下降很明显,明暗适应的视网膜电图很大程度上降低或者不存在,也即a、b波延迟、低平或消失[17]。白内障可能在RP的中期出现,眼科检查发现,视野缩小,但是中心视野在早期保持正常,至到疾病的晚期完全缺失。有些并发黄斑囊样水肿和黄斑前膜,而PDE6b基因缺失引起的视网膜色素变性很少引起黄斑前膜[18]。RP伴有很多疾病称为RP综合征,其中常染色体隐性遗传RP占到RP总数的20%~25%[19],到目前为止,由PDE6b基因突变所造成的RP1%~2%,并以常染色体隐性遗传为主,由这个基因突变所引起的RP占arRP患者的4%~5%。超过35个基因座及基因被认为与常染色体隐性遗传的RP相关。PDE的异常在很多视网膜色素变性疾病患者中广泛存在,研究证实PDE活性的丧失、cGMP积聚和光感受器的凋亡同时存在。
根据人类基因突变库,到目前为止PDE6b至少有24种突变可能引起视网膜色素变性,包括C270X,Q298X,L527P,R531X,H557Y,G576D,H620(1-bp del),K706X,L854V,W807R,I535N,R552Q,P387L,D600N,H258N,L228I,E640fs,L669R,W378,R799X,H337R,R560C和两个剪接突变P496(1-bp del)和P2193(1+bp.del)[20]等。大部分的突变类型是错义突变,尽管这种突变类型对于下游基因编码的影响还不是很清楚,但它们对酶功能的影响还是大于无义突变所造成的影响。人类的PDE6b基因的多数突变是在常染色体隐性遗传的视网膜色素变性患者中发现的。
PDE6b基因突变类型有所不同,其中点突变分为碱基替换和移码突变,碱基替换分为密码子区域的替换和非密码子区域的替换,密码子突变类型又分为同义突变、错义突变、无义突变和终止密码子突变。
其中密码子区域的突变位点如下。错义突变:L527P和H557Y发生在PDE6b基因的同系物催化区[21];I535N(c.1604T>A)处发生了一个错义突变,另外一个在H557Y(c.1699C>T)发生的错义突变[22];W807R(c.2419T>A),21号外显子密码子2419发生错义突变,也就是在位置807上一个A替代了T,最终导致活性高的精氨酸代替了保守的色氨酸,尽管这个突变发生PDE蛋白的催化区以外,但它发生的位点却是一个高度保守的位置[15];R552Q(c.1655G>A)和P387L(c.1160C>T),前者在密码子552位一个谷氨酸代替了精氨酸,后者在密码子387位上亮氨酸取代了脯氨酸,通过单倍体连锁分析显示PDE6b基因的位于4号染色体上,这两个突变的位置都影响了PDE蛋白的三维立体结构[23];另外外显子12位发生I533N(c.1604T>A)和在外显子13位上发生的H557Y(c.1669C>T)[17],R560C(c.1678C>T)发生在外显子13位的点突变。Jacobson等[24]曾经报导了一例因C270X的突变缺乏PDE6β患者,造成视网膜外周增厚而中央凹处变薄;Ullah等[25]从350多个视网膜色素变性的家系中筛选出11个家族存在PDE6基因缺失,PDE6b基因缺失c.243delG (p.R82Afs68*)和c.12_15delTGAG (p.S4Rfs23*)与视网膜色素相关。无义突变:三个无义突变Gln-298-X、Arg-531-X、Lys-706-X[16,21],其中Lys-706-X突变会引起PDE6B蛋白一段催化区的缺失[26]。Cheng等[20]在对144个基因相关的视网膜病变进行研究时发现,两个新的突变基因,分别是在外显子9位发生的W378X(c.1133G>A)突变和在外显子21位发生的R799X(c.2395C>T)突变,这两个突变区域皆位于PDE6b的保守区域,经证实复合杂合的PDE6b基因的两个无义突变也能造成RP。外显子12位的无义突变,即Met523Arg(c.1568T>G)[27]。终止密码子突变:Shen等[28]通过对一组视网膜色素变性的家庭进行新一代外显子基因测序分析,得到一个新的PDE6b基因突变类型纯合子c.1923_1969,这个突变位点位于外显子16位,有一个47bp的插入和一个6bp的删除,最终造成在641位氨基酸发生移码突变,并且还提前出现了一个终止密码子,外显子16~22位的丢失引起蛋白质缩短,在氨基酸555-792的位置打断了cGMP水解的催化区域,而这个终止密码子很大可能导致PDE6β失去功能,引起高水平的cGMP,随后通道关闭的数量减少。影响非密码子区域替换如下。外显子剪接突变:Mclaughlin等[21]在遗传的视网膜色素变性的家族遗传谱中发现一处PDE6b基因的剪接突变,引起视网膜外核层PDE的减少,cGMP的聚积;Beheshtian等[29]对13个家族的10个家庭进行了arRP的基因突变点的检测,在PDE6b基因的外显子8位一处剪接突变。
移码突变:Michael等[20]在外显子15位发生的一个碱基缺失H620(1-bp del)造成移码突变;内含子2位AG转换为AT引起外显子3位一个阅读框的缺失造成移码突变[30]。
3 展 望
PDE6b基因的表达异常是常染色体隐性遗传arRP首要致病基因,RP在西方发病率为1/4 000-1/3 500[31],国内统计发病率为1/3784,全世界约有150万以上患者,至今尚无有效防治手段。目前为止,有144个基因,超过4 000个突变和视网膜疾病明确相关。RP是一组同源性较高的一组视网膜疾病,有不同的表型涉及到基因突变的多样性。因此通过对PDE6b基因的研究,且已掌握此基因的定位、功能和突变类型。然而,PDE6b基因异常致病的分子机制及基因的表达表型的影响还不是很清楚,有待研究的因素还很多。尽管所有自然突变形成的小鼠模型和人类因PDE6b基因缺陷所导致的视网膜色素变性疾病有相似的特点,但治疗方面的发现还不能充分满足人类疾病的需要。对于PDE6b基因缺陷的人体研究还比较少,随着更多临床前期和临床研究,基因替代治疗使用玻璃体内注射高效渗透性强的AAV载体,结合抗氧化类药物、生长因子、或者其它药物变成更重要的措施治疗常染色体隐性遗传视网膜色素变性。这篇综述基于对小鼠PDE6b基因的研究以及部分RP患病家族的研究,为人体研究提供了依据,并对RP患者治疗提供了依据。
[1] KEELER C E. The inheritance of a retinal abnormality in white mice [J]. Proc Natl Acad Sci U S A,1924,10(7): 329-333.
[2] PITTLER S J, BAEHR W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse [J]. Proc Natl Acad Sci U S A,1991, 88(19): 8322-8326.
[3] PITTLER S J, COAN P, KONTZEN P L, et al. Aminoglycosides partially restore photoreceptor function in the rd1 mouse [J]. Invest Ophthalmol Vis Sci,2001,42(4): S528-S528.
[4] BOWES C, LI T, DANCIGER M, et al. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase [J].Nature,1990, 347(6294): 677-680.
[5] CHANG B, HAWES N L, HURD R E, et al. Retinal degeneration mutants in the mouse [J].Vision Res,2002, 42(4): 517-525.
[6] HART A W, MCKIE L, MORGAN J E, et al. Genotype-phenotype correlation of mouse pde6b mutations [J].Invest Ophthalmol Vis Sci,2005, 46(9): 3443-3450.
[7] GARGINI C, TERZIBASI E, MAZZONI F, et al. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study [J].J Comp Neurol,2007, 500(2): 222-238.
[8] CHANG B, HAWES N L, PARDUE M T, et al. Two mouse retinal degenerations caused by missense mutations in the β-subunit of rod cGMP phosphodiesterase gene [J].Vision Res,2007, 47(5): 624-633.
[9] AQUIRRE G, FARBER D, LOLLEY R, et al. Rod-cone dysplasia in Irish setters: a defect in cyclic GMP metabolism in visual cells[J].Science,1978,201(4361):1133-1134.
[10] AGUIRRE G D, RUBIN L F.Rod-cone dysplasia (progressive retinal atrophy) in Irish setters[J].J Am Vet Med Assoc,1975. 166(2). 157-164.
[11] SUBER M L, PITTLER S J, QIN N, et al. Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase beta-subunit gene [J].Proc Natl Acad Sci U S A,1993,90(9):3968-3972.
[12] DEKOMIEN G, RUNTE M, GÖDDE R, et al. Generalized progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene [J].Cytogenet Cell Genet,2000,90(3-4):261-267.
[13] GOLDSTEIN O, MEZEY J G, SCHWEITZER P A, et al. IQCB1 and PDE6B mutations cause similar early onset retinal degenerations in two closely related terrier dog breeds [J].Invest Ophthalmol Vis Sci,2013, 54(10): 7005-7019.
[14] LEM J, FAIN G L. Constitutive opsin signaling: night blindness or retinal degeneration? [J].Trends Mol Med,2004, 10(4): 150-157.
[15] HMANI-AIFA M, BENZINA Z, ZULFIQAR F, et al. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family [J].Eur J Hum Genet,2009, 17(4): 474-482.
[16] MCLAUGHLIN M E, SANDBERG M A, BERSON E L, et al. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa [J].Nat Genet,1993, 4(2): 130-134.
[17] KUNIYOSHI K, SAKURAMOTO H, YOSHITAKE K, et al. Reduced rod electroretinograms in carrier parents of two Japanese siblings with autosomal recessive retinitis pigmentosa associated with PDE6B gene mutations [J].Doc Ophthalmol,2015, 131(1): 71-79.
[18] OGINO K, OISHI A, OISHI M, et al. Efficacy of column scatter plots for presenting retinitis pigmentosa phenotypes in a Japanese cohort [J].Transl Vis Sci Technol,2016, 5(2): 4.
[19] DAIGER S P, SULLIVAN L S, BOWNE S J. Genes and mutations causing retinitis pigmentosa [J]. Clin Genet,2013, 84(2): 132-141.
[20] CHENG L L, HAN R Y, YANG F Y, et al. Novel mutations in PDE6B causing human retinitis pigmentosa [J].Int J Ophthalmol,2016,9(8):1094-1099.
[21] MCLAUGHLIN M E, EHRHART T L, BERSON E L, et al. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa [J].Proc Natl Acad Sci U S A,1995, 92(8): 3249-3253.
[22] SAGA M, MASHIMA Y, AKEO K, et al. A novel homozygous Ile535Asn mutation in the rod cGMP phosphodiesterase beta-subunit gene in two brothers of a Japanese family with autosomal recessive retinitis pigmentosa [J].Curr Eye Res,1998, 17(3): 332-335.
[23] ALI S, RIAZUDDIN S A, SHAHZADI A, et al. Mutations in the beta-subunit of rod phosphodiesterase identified in consanguineous Pakistani families with autosomal recessive retinitis pigmentosa [J].Mol Vis,2011, 17: 1373-1380.
[24] JACOBSON S G, SUMAROKA A, ALEMAN T S, et al. Evidence for retinal remodelling in retinitis pigmentosa caused by PDE6B mutation [J].Br J Ophthalmol,2007, 91(5): 699-701.
[25] ULLAH I, KABIR F, GOTTSCH C B, et al. Mutations in phosphodiesterase 6 identified in familial cases of retinitis pigmentosa [J].Hum Genome Var,2016, 3: 16036-16039.
[26] BISWAS P, DUNCAN J L, MARANHAO B, et al. Genetic analysis of ten pedigrees with inherited retinal degeneration (IRD) by exome sequencing and phenotype-genotype association [J]. Physiol Genomics,2017, 49(4):216-229.
[27] BOCQUET B, MARZOUKA N A, HEBRARD M, et al. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations [J].Mol Vis,2013, 19: 2487-2500.
[28] SHEN S, SUJIRAKUL T, TSANG S H. Next-generation sequencing revealed a novel mutation in the gene encoding the beta subunit of rod phosphodiesterase [J].Ophthalmic Genet,2014, 35(3): 142-150.
[29] BEHESHTIAN M, SAEE RAD S, BABANEJAD M, et al. Impact of whole exome sequencing among Iranian patients with autosomal recessive retinitis pigmentosa [J].Arch Iran Med,2015, 18(11): 776-785.
[30] DANCIGER M, BLANEY J, GAO Y Q, et al. Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa [J].Genomics,1995,30(1):1-7.
[31] RIVOLTA C, SHARON D, DEANGELIS M M, et al. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns [J].Hum Mol Genet, 2002,11(10):1219-1227.
[本文编辑] 姬静芳
The role of abnormal expression of the PDE6b gene in retinitis pigmentosa
YUAN Li1, LIU Yan1*, LIU Te2
1. Department of Ophthalmology, Shanghai General Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200080, China2. Shanghai Geriatric Institute of Traditional Chinese Medicine, Shanghai 200031, China
Phosphodiesterases (PDEs) are a large family of proteins divided into 11 subfamilies, namely PDE1-PDE11. PDE6 is a PDEs subfamily, the PDE protein encoded by the PDE6 gene, which hydrolyses cGMP, the specific receptor of rod outer segment membrane disc ion channels and also an important molecule in the vertebrate photoreceptor cells that converts light signals into electrical signals. The deletion of pde6b gene results in a nonfunctional PDE and an accumulation of cGMP in the photoreceptor cells, causing the increase of cationic photoreceptor cells, cell poisoning and cell death. The photoreceptor cells in the retina constantly deteriorate, indirectly affecting the retina converting the light signals into electrical signals. The abnormal expression of pde6b gene is one of the most important causes of autosomal recessive retinitis pigmentosa. Therefore, the abnormal expression of pde6b gene in animal models and familial inheritance has gradually become the focus of research in recent years. This paper focuses on the structure, function, and mutation types of pde6b gene, and their influence on the corresponding structure and function of retinitis pigmentosa.
pde6b; retina; rd1; gene deletion; retinitis pigmentosa
2016-11-01 [接受日期] 2017-04-13
国家自然科学基金面上项目(81371068). Supported by General Fund of National Natural Science Foundation of China (81371068).
袁 莉,硕士生. E-mail: yuanli0720@163.com
*通信作者(Corresponding author). Tel: 021- 56663031, E-mail: yanliu201410@126.com
10.12025/j.issn.1008-6358.2017.20161014
R 774.1+3
A
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
广西医科大学学报(2022年5期)2022-06-07
中国循证儿科杂志(2022年2期)2022-05-26
昆明医科大学学报(2022年3期)2022-04-19
福建农业学报(2021年6期)2021-08-18
中国生殖健康(2020年4期)2021-01-18
教学考试(高考生物)(2020年4期)2020-11-18
生物学通报(2020年11期)2020-10-22
中南医学科学杂志(2019年6期)2019-12-05
发明与创新·中学生(2019年6期)2019-06-26
