
缺血性心肌病心室肌细胞电重构的研究现状
2017-07-09 12:38:30李小龙万征
中华心脏与心律电子杂志 2017年3期
李小龙 万征
慢性心力衰竭(简称心衰)患者常伴发各种室性心律失常,其死亡率约占心衰患者总死亡率的13%;而随着冠状动脉性心脏病(简称冠心病)发病率和心脏重构的增加,缺血性心肌病(Ischemic Cardiomyopathy,ICM)已逐渐成为心衰的主要病因[1-2]。心脏重构是心脏在病理环境中的一种适应性改变,包括心肌重构、血管重构、能量代谢重构和电重构等[3-4]。而其中电重构又是心律失常的基础,通常发生在心衰早期,从细胞层面开始逐渐累及整个心脏。近年来,心脏电重构已成为ICM和心衰的研究焦点。
一、心室肌细胞
心衰时心室肌细胞的适应性变化会导致细胞电生理特性的改变,即细胞电重构。其中,离子通道重构是主体,包括膜电流、通道蛋白表达和通道调节的异常改变等,这将引起细胞动作电位(Action Potential,AP)的改变,如细胞动作电位时程(Action Potential Duration,APD)延长等,这是心律失常发生的直接原因之一。见图1。
比较正常心室肌细胞,衰竭心脏心室肌细胞APD明显延长[5]。

图1 来自正常和衰竭心脏的心室肌细胞AP示意图
1.离子通道和膜电流的重构:在心衰模型中,心肌细胞的电生理特征主要表现为可记录到的APD延长[6],涉及到多种离子通道重构和膜电流变化(表1)。在ICM中,左室心肌细胞快钠电流(Transient Sodium Current,INaT)在缺血时明显减少[7],而INaT的减少将导致AP 0期上升速率降低,使兴奋传导速度减慢,进而使再灌注区受损或正在恢复功能的细胞和正常细胞间兴奋的传导速度产生差异,从而诱发”折返”等异常电生理现象。同时,由于晚钠电流(Late Sodium Current,INaL)参与AP平台期的形成,且当发生ICM或心衰时,INaL明显增大[8],从而使平台期显著延长。在心衰模型中,内向整流钾电流(Inwardly Rectifying Potassium Current,Ik1)减少,延长复极,降低复极后不应性,从而易诱发迟发后除极(Delayed Afterdepolarizations,DADs)和功能性折返[9]。
近年来,一些新发现的离子通道也逐渐成为治疗和延缓心衰进展的研究热点之一,如ATP敏感性钾(ATP-sensitive Potassium,KATP)通道、小电导钙激活钾(Small Conductance Calcium-activated Potassium,SK)通道以及双孔钾(two-pore-domain potassium,K2P)通道。在正常心脏中,细胞膜上KATP通道的高表达有助于心肌细胞快速复极,在机体耗氧量增多的情况下,有助于增加心率和心输出量;K2P通道也是促进心肌细胞复极,稳定细胞膜电位[10]。而SK通道在正常心脏中的表达量很少,因此在正常生理状态下,其并不会对心室肌细胞的电活动产生较大影响[11]。在心衰模型中,KATP通道的表达量减少了35%~40%,使心肌细胞复极减慢,在心率加快的情况下,易引起心肌细胞耗氧量明显增加,导致了心肌细胞氧需求与氧供应之间的不平衡,引起心肌缺血性损伤及心功能的进一步失调[12],进而加速心衰进展,加重电生理紊乱,引起恶性循环;K2P2.1通道在心衰时表达减少,易引起心室电传导紊乱及室性心律失常,但其机制尚未明确[13];而SK通道同样可以诱发室性心律失常。有研究认为在衰竭心室肌细胞中,ISK增多[14];同时, 单相动作电位复极90%的时程(Monophasic Action Potential Duration at 90% Repolarization,MAPD90) 缩短,并表现出致室性心律失常作用;而当ISK被阻滞时,缩短的MAPD90会恢复正常,产生心脏保护作用[15]。
2.离子通道与转运体的重构:少量的Ca2+通过L型钙通道(LTCC)进入细胞内,并刺激兰尼碱受体(RyRs),使RyR2开放,进而使肌浆网释放大量Ca2+进入细胞质,并刺激肌丝蛋白,从而引发收缩活动。通过关闭L型钙通道(L-type Ca2+channel,LTCC)和兰尼碱受体(Ryanodine Receptors,RyRs),肌浆网钙

表1 心衰模型心室肌不同通道电流变化概况
注:“—”为无明显变化;“↑”为延长;“↓”为缩短;“Ik1”为内向整流钾电流;“Ito”为瞬时外向钾电流;“Ik”为延迟整流钾电流;“INaL”为晚钠电流;“ICa-L”为L型钙电流;“IKATP”为ATP敏感性钾电流;“IKCa”钙激活钾电电流。
ATP酶(SERCA)回摄Ca2+及通过钠钙交换体(Na+/Ca2+Exchanger,NCX)的Ca2+流出,从而建立细胞质低浓度Ca2+环境,引发舒张活动。
近年来研究显示离子通道电流的改变通常提示通道蛋白及构象方面的重构,在这一层面的研究和认识逐步深入。
在正常心脏中,Ca2+信号(Ca2+signaling)是维持心脏正常电活动和收缩舒张功能的核心环节之一。在该环节中,由心肌细胞细胞膜上和细胞内的离子通道和转运体共同参与,如钠钙交换体(Na+/Ca2+exchanger,NCX)和兰尼碱受体(Ryanodine Receptors,RyRs)等,它们在电传导、兴奋收缩耦联等方面发挥重要作用。见图2。钙调蛋白激酶Ⅱ(Ca2+/Calmodulin-dependent Protein Kinase Ⅱ,CaMKⅡ)则在其中居重要地位,在ICM所致的心衰中,CaMKⅡ的表达量及活性均明显增加,使得RyRs发生过磷酸化,肌浆网Ca2+泄露增加[31],诱发钙超载,激活NCX并产生内向电流,引发DADs,进而触发AP及室性心律失常。近期有研究表明可通过阻滞NOD1(Nucleotide-binding Oligomerization Domain-containing Protein 1)信号通路来减少异常收缩期Ca2+释放和RyRs的过磷酸化来减少上述Ca2+调控异常的发生[32],以减少NCX激活所介导的内向电流的产生,进而减少AP的触发及心律失常的发生。在生理条件下,NCX1(Na+/Ca2+exchanger 1,NCX1)是钙稳态必需的调节因子,其表达增加可以减弱压力过载介导的心脏结构重构[33]。因此,NCX1可能是延缓心衰进展的潜在治疗靶点。
有研究报道称CaMKⅡ抑制剂SMP-114主要通过减少Ca2+火花(Ca2+sparks)的发生,从而使Ca2+泄露明显减少以维持钙稳态,且可明显减少INaL[34],进而减少室性心律失常的发生。在ICM,尽管Ca2+火花的发生减少,但RyRs对Ca2+敏感性增强,促进Ca2+火花向Ca2+波(Ca2+waves)的扩展,使Ca2+波的发生频率增加,产生致心律失常作用[33]。此外,钾离子相互作用蛋白在心衰时表达减少,引起L型钙电流增大,延长APD,同时明显减少钙瞬变幅度及延长瞬变时间[33],进而诱发早发后除极和(或)DADs,形成了心律失常的电生理基础。

图2 钙调蛋白与T管及肌浆网的关系模型图[30]
二、细胞外基质和细胞间连接
在衰竭心脏中,心室肌的细胞构成和细胞外基质(extracellular matrix,ECM)的改变、缝隙连接的表达和分布异常,将引起细胞间电传导速度和路径的改变,增加电传导的各向异性(anisotropic),构成了各种折返性心律失常的电生理基础(图3)。
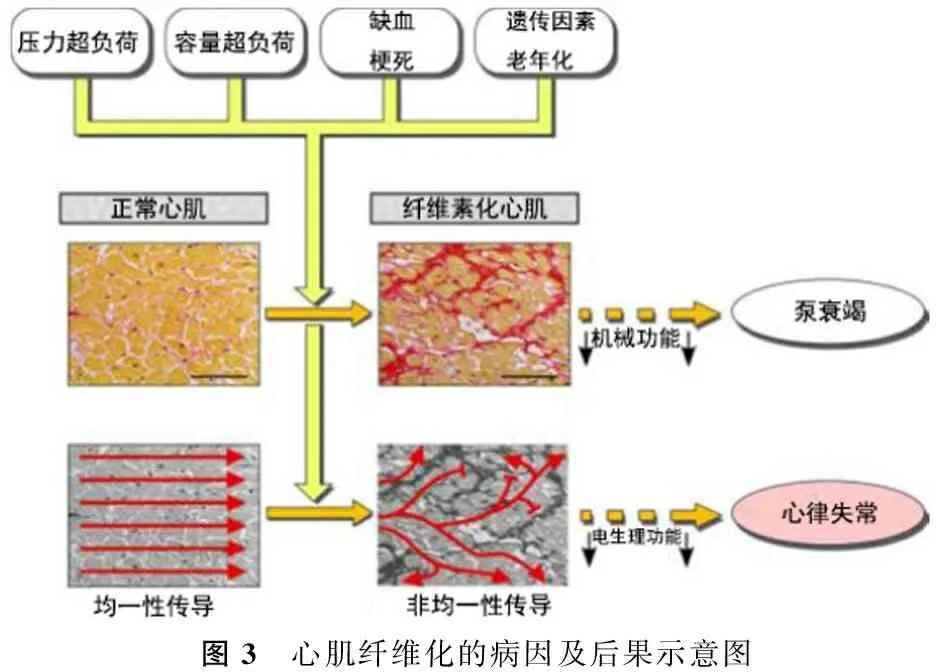
图3 心肌纤维化的病因及后果示意图
显微图像显示结缔组织(胶原:红色)在正常心肌(左)和广泛左心室纤维化患者心肌(右)的分布情况[35]。比例尺=100 μm。
1.细胞外基质:衰竭的心脏可发生一系列引起电生理异常的病理改变,包括心肌肌束间及血管周围胶原纤维显著增生,可见灶性纤维瘢痕,心肌细胞萎缩或坏死、排列紊乱和间质血管扩张充血,使得细胞外基质(Extracellular Matrix,ECM)发生重度重构[36]。
在衰竭心脏心室成纤维细胞中,SOCE/CRAC通道(Store-operated Ca2+Entry/Ca2+Release-activated Channels)介导的Ca2+电流明显增大,且该通道成孔亚单位ORAI表达增多,可导致衰竭心脏胶原蛋白分泌增多[37];同时,ECM中收缩蛋白的表达增多[38],二者共同促进心肌间质纤维化;此外,过量的ECM蛋白可阻断心肌细胞束的连续性,而这会使心肌电传导的各向异性增加,导致局部电传导紊乱和折返性心律失常[39](图4)。目前,有研究人员发现可以通过抑制G蛋白βγ亚单位及其与G蛋白耦联受体激酶2之间的联系来消除上述病理性心肌纤维化[40],进而改善心脏结构重构和电重构,延缓心衰进展,减少心律失常的发生;此外,有研究报道,在心衰时心脏赖氨酰氧化酶的表达量及活性均进行性增加[41]以及心脏成纤维细胞特异激活转录因子3的表达上调[42],均可削弱心肌纤维化。
(a)正常心肌组织。(b)与心肌细胞束平行的纤维化(如发生反应性纤维化)。纵向细胞-细胞连接完整,且纵向传导不受影响。(c)平行纤维化和穿过心肌束的纤维化(如发生梗死后修复性纤维化)[43]。由于死亡的心肌细胞被成纤维细胞和纤维化细胞外基质蛋白替换而产生的物理分离,导致纵向传导的局部异常。
2.细胞间连接:在正常心脏中,缝隙连接是心肌细胞之间连接的主要方式,而在心室肌中,缝隙连接蛋白主要为Cx43。有报道ICM患者的心室肌组织中,Cx43的数量明显减少[44],且从闰盘到细胞边界重新分布,由细胞的端-端连接转变为侧-侧连接,偏侧化增加,且大部分Cx43发生脱磷酸化改变,导致细胞间兴奋耦联下降[45]。这些异常都可导致心肌发生不均一性电传导改变,使心肌细胞间传导速度变慢,APD延长,形成折返环路等,从而诱发恶性室性心律失常[46]。此外,Cx43的重构可能也会导致心肌细胞内钙超载与ATP浓度降低[47],影响心室肌兴奋收缩耦联。因此,抑制Cx43重构,可以恢复心肌细胞间电耦联,改善冲动在细胞间传导,使心脏电活动趋于同步,减少心律失常发生。
此外,心肌ECM中的整合素、基质细胞蛋白和金属蛋白酶在胞间连接中同样发挥着重要作用[48],可以作为治疗心脏重构和心律失常的直接靶点。
三、展望
目前,随着分子生物学的进展,心衰研究已进入分子时代,着眼于衰竭细胞中离子通道亚单位及相关构型构象等,从而在分子水平阐述电重构,可以全方位立体化解读ICM的电重构现象,进而为ICM合并心衰患者心律失常的治疗提供新思路,为研发分子靶向药物提供新方向,提供心衰患者心律失常“上游治疗”基础。
[1] 苏立杰,李觉,胡大一.心力衰竭的研究现状和进展[J].中国心血管病研究杂志,2009,7(3):222-225.
[2] 顾东风,黄广勇,何江,等.中国心力衰竭流行病学调查及其患病率[J].中华心血管病杂志,2003,31(1):3-6.
[3] Heusch G,Libby P,Gersh B,et al.Cardiovascular remodelling in coronary artery disease and heart failure[J].Lancet,2014,383(9932):1933-1943.
[4] Armoundas AA,Wu R,Juang G,et al.Electrical and structural remodeling of the failing ventricle[J].Pharmacol Ther,2001,92(2-3):213-230.
[5] Rose J,Armoundas AA,Tian Y,et al.Molecular correlates of altered expression of potassium currents in failing rabbit myocardium[J].Am J Physiol Heart Circ Physiol,2005,288(5):H2077-2087.
[6] Benz A,Kossack M,Auth D,et al.miR-19b Regulates Ventricular Action Potential Duration in Zebrafish[J].Sci Rep,2016,6:36033.
[7] 王芳,李洪仕,万征,等.阿托伐他汀对大鼠缺血再灌注左室心肌细胞瞬时钠电流的作用[J].中华心脏与心律电子杂志,2014,2(1):41-44.
[8] Bengel P,Ahmad S,Sossalla S.Inhibition of Late Sodium Current as an Innovative Antiarrhythmic Strategy[J].Curr Heart Fail Rep,2017,14(3):179-186.
[9] Klein MG,Shou M,Stohlman J,et al.Role of suppression of the inward rectifier current in terminal action potential repolarization in the failing heart[J].Heart Rhythm,2017,14(8):1217-1223.
[10] Feliciangeli S,Chatelain FC,Bichet D,et al.The family of K2P channels:salient structural and functional properties[J].J Physiol,2015,593(12):2587-2603.
[11] Hundahl LA,Sattler SM,Skibsbye L,et al.Pharmacological blockade of small conductance Ca2+-activated K+channels by ICA reduces arrhythmic load in rats with acute myocardial infarction[J].Pflugers Arch,2017,469(5-6):739-750.
[12] Gao Z,Sierra A,Zhu Z,et al.Loss of ATP-Sensitive Potassium Channel Surface Expression in Heart Failure Underlies Dysregulation of Action Potential Duration and Myocardial Vulnerability to Injury[J].PLoS One,2016,11(3):e0151337.
[13] Wiedmann F,Schmidt C,Lugenbiel P,et al.Therapeutic targeting of two-pore-domain potassium (K(2P)) channels in the cardiovascular system[J].Clin Sci (Lond),2016,130(9):643-650.
[14] Chua SK,Chang PC,Maruyama M,et al.Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles[J].Circ Res,2011,108(8):971-979.
[15] Gui L,Bao Z,Jia Y,et al.Ventricular tachyarrhythmias in rats with acute myocardial infarction involves activation of small-conductance Ca2+-activated K+ channels[J].Am J Physiol Heart Circ Physiol,2013,304(1):H118-130.
[16] Fernández-Velasco M,Ruiz-Hurtado G,Delgado C.I K1 and I f in ventricular myocytes isolated from control and hypertrophied rat hearts[J].Pflugers Arch,2006,452(2):146-154.
[17] Li GR,Lau CP,Ducharme A,et al.Transmural action potential and ionic current remodeling in ventricles of failing canine hearts[J].Am J Physiol Heart Circ Physiol,2002,283(3):H1031-1041.
[18] Pogwizd SM,Schlotthauer K,Li L,et al.Arrhythmogenesis and contractile dysfunction in heart failure:Roles of sodium-calcium exchange,inward rectifier potassium current,and residual beta-adrenergic responsiveness[J].Circ Res,2001,88(11):1159-1167.
[19] Ufret-Vincenty CA,Baro DJ,Lederer WJ,et al.Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure[J].J Biol Chem,2001,276(30):28197-28203.
[20] Hardziyenka M,Campian ME,Verkerk AO,et al.Electrophysiologic remodeling of the left ventricle in pressure overload-induced right ventricular failure[J].J Am Coll Cardiol,2012,59(24):2193-2202.
[21] 王国芹,李洪仕,万征,等.阿托伐他汀对大鼠缺血再灌注左室心肌细胞瞬时外向钾电流的作用[J].中华心脏与心律电子杂志,2013,1(1):43-46.
[22] Cooper PJ,Soeller C,Cannell MB.Excitation-contraction coupling in human heart failure examined by action potential clamp in rat cardiac myocytes[J].J Mol Cell Cardiol,2010,49(6):911-917.
[23] Xu X,Salata JJ,Wang J,et al.Increasing I(Ks) corrects abnormal repolarization in rabbit models of acquired LQT2 and ventricular hypertrophy[J].Am J Physiol Heart Circ Physiol,2002,283(2):H664-670.
[24] Rodrigues JLF,de Azevedo Carvalho AC,Pimentel EB,et al.Chronic enalapril treatment increases transient outward potassium current in cardiomyocytes isolated from right ventricle of spontaneously hypertensive rats[J].Naunyn Schmiedebergs Arch Pharmacol,2017,390(3):225-234.
[25] Xi Y,Wu G,Yang L,et al.Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload model[J].Eur J Heart Fail,2009,11(8):749-757.
[26] Markandeya YS,Tsubouchi T,Hacker TA,et al.Inhibition of late sodium current attenuates ionic arrhythmia mechanism in ventricular myocytes expressing LaminA-N195K mutation[J].Heart Rhythm,2016,13(11):2228-2236.
[27] Chi L,Belardinelli L,Zeng A,et al.Inhibition of late Na+ current,a novel target to improve diastolic function and electrical abnormalities in Dahl salt-sensitive rats[J].Am J Physiol Heart Circ Physiol,2016,310(10):H1313-1320.
[28] Iyer V,Heller V,Armoundas AA.Altered spatial calcium regulation enhances electrical heterogeneity in the failing canine left ventricle:implications for electrical instability[J].J Appl Physiol (1985),2012,112(6):944-955.
[29] Ortega A,Tarazón E,Roselló-Lletí E,et al.Patients with Dilated Cardiomyopathy and Sustained Monomorphic Ventricular Tachycardia Show Up-Regulation of KCNN3 and KCNJ2 Genes and CACNG8-Linked Left Ventricular Dysfunction[J].PLoS One,2015,10(12):e0145518.
[30] Houser SR,Molkentin JD.Does contractile Ca2+control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes?[J].Sci Signal,2008,1(25):pe31.
[31] Fischer TH,Eiringhaus J,Dybkova N,et al.Ca2+/calmodulin-dependent protein kinase Ⅱ equally induces sarcoplasmic reticulum Ca2+leak in human ischaemic and dilated cardiomyopathy[J].Eur J Heart Fail,2014,16(12):1292-1300.
[32] Val-Blasco A,Piedras MJ,Ruiz-Hurtado G,et al.Role of NOD1 in Heart Failure Progression via Regulation of Ca2+Handling[J].J Am Coll Cardiol,2017,69(4):423-433.
[33] Ujihara Y,Iwasaki K,Takatsu S,et al.Induced NCX1 overexpression attenuates pressure overload-induced pathological cardiac remodelling[J].Cardiovasc Res,2016,111(4):348-361.
[34] Neef S,Mann C,Zwenger A,et al.Reduction of SR Ca2+leak and arrhythmogenic cellular correlates by SMP-114,a novel CaMKⅡ inhibitor with oral bioavailability[J].Basic Res Cardiol,2017,112(4):45.
[35] Rohr S.Myofibroblasts in diseased hearts:new players in cardiac arrhythmias?[J].Heart Rhythm,2009,6(6):848-856.
[36] 贾政,魏玲,刘茜,等.重组腺病毒介导Klotho基因转导对大鼠心力衰竭心肌重构的影响[J].中华心血管病杂志,2015,43(3):219-226.
[37] Ross GR,Bajwa T,Edwards S,et al.Enhanced store-operated Ca2+influx and ORAI1 expression in ventricular fibroblasts from human failing heart[J].Biol Open,2017,6(3):326-332.
[38] DeAguero JL,McKown EN,Zhang L,et al.Altered protein levels in the isolated extracellular matrix of failing human hearts with dilated cardiomyopathy[J].Cardiovasc Pathol,2017,26:12-20.
[39] Yue L,Xie J,Nattel S.Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation[J].Cardiovasc Res,2011,89(4):744-753.
[40] Travers JG,Kamal FA,Valiente-Alandi I,et al.Pharmacological and Activated Fibroblast Targeting of Gβγ-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression[J].J Am Coll Cardiol,2017,70(8):958-971.
[41] El HEC,El HMC,Ninh VK,et al.Detrimental role of lysyl oxidase in cardiac remodeling[J].J Mol Cell Cardiol,2017,109:17-26.
[42] Li Y,Liu B,Liang C.Letter by Li et al Regarding Article,"Cardiac Fibroblast-Specific Activating Transcription Factor 3 Protects Against Heart Failure by Suppressing MAP2K3-p38 Signaling"[J].Circulation,2017,136(21):2092-2093.
[43] Yue L,Xie J,Nattel S.Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation[J].Cardiovasc Res,2011,89(4):744-753.
[44] Nikolaidou T,Cai XJ,Stephenson RS,et al.Congestive Heart Failure Leads to Prolongation of the PR Interval and Atrioventricular Junction Enlargement and Ion Channel Remodelling in the Rabbit[J].PLoS One,2015,10(10):e0141452.
[45] 臧小彪,张荣峰.心力衰竭对缝隙连接蛋白的影响[J].中国心脏起搏与心电生理杂志,2012,26(1):83-85.
[46] Louzao-Martinez L,Vink A,Harakalova M,et al.Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy[J].Int J Cardiol,2016,220:634-646.
[47] Gao J,Zhao Y,Wang Y,et al.Anti-arrhythmic effect of acupuncture pretreatment in the rats subjected to simulative global ischemia and reperfusion--involvement of intracellular Ca2+and connexin 43[J].BMC Complement Altern Med,2015,15:5.
[48] Valiente-Alandi I,Schafer AE,Blaxall BC.Extracellular matrix-mediated cellular communication in the heart[J].J Mol Cell Cardiol,2016,91:228-237.
李小龙,万征.缺血性心肌病心室肌细胞电重构的研究现状[J/CD].中华心脏与心律电子杂志,2017,5(3):178-182.
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:16:04
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:24
小学科学(学生版)(2019年10期)2019-11-16 08:55:04
中国环境监察(2017年5期)2017-10-23 05:26:48
海南医学(2016年8期)2016-06-08 05:43:00
电测与仪表(2016年14期)2016-04-11 12:34:22
淮海医药(2015年2期)2016-01-12 04:33:21
中国病理生理杂志(2015年8期)2015-12-21 12:38:08
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国火炬(2014年6期)2014-07-24 14:16:34
