
HPLC法测定维格列汀片中的有关物质
2017-07-07 15:09张梦泽河南省口岸食品检验检测所郑州450003河南省食品药品检验所郑州450003
中国药房 2017年15期
黄 姗,张梦泽(1.河南省口岸食品检验检测所,郑州 450003;.河南省食品药品检验所,郑州 450003)
HPLC法测定维格列汀片中的有关物质
黄 姗1*,张梦泽2(1.河南省口岸食品检验检测所,郑州 450003;2.河南省食品药品检验所,郑州 450003)
目的:建立测定维格列汀片中有关物质的方法。方法:采用高效液相色谱法。色谱柱为XterraMSC18,流动相为[磷酸盐缓冲液-水-乙腈-甲醇(400∶600∶15∶15,V/V/V/V)]-[磷酸盐缓冲液-乙腈-甲醇(400∶450∶150,V/V/V)](梯度洗脱),流速为1.2m L/m in,检测波长为210 nm,柱温为35℃,进样量为10µL。结果:杂质A、B、C、D检测质量浓度线性范围分别为18.80~188.0µg/m L(r=0.999 5)、22.64~226.4µg/m L(r=0.999 6)、21.74~217.4µg/m L(r=0.999 7)、19.12~191.2µg/m L(r=0.999 8);检测限分别为4.18、2.68、1.12、1.34µg/m L;精密度、稳定性、重复性试验的RSD<3%;加样回收率分别为97.9%~103.1%(RSD=2.01%,n=9)、98.8%~104.1%(RSD=1.93%,n=9)、98.0%~103.6%(RSD=1.81%,n=9)、100.8%~104.1%(RSD=0.98%,n=9)。结论:该方法操作简单、结果准确,可用于维格列汀片中有关物质的测定。
维格列汀片;有关物质;高效液相色谱法
维格列汀(Vildagliptin)化学名为1-{[(3-羟基-l-金刚烷基)氨基]乙酰基}-2-氰基-(S)-四氢吡咯烷,是一种具有选择性和口服活性的特异性二肽酰肽酶-4(DPP-4)抑制剂[1],可以刺激胰岛素基因表达和胰岛素生物合成,属于新一代口服降血糖药物[2-4]。目前,我国关于维格列汀有关物质的测定尚未见报道[5]。为更好地控制维格列汀片的质量,笔者采用高效液相色谱法(HPLC)建立了测定该制剂中有关物质的方法。
1 材料
1.1 仪器
1260型HPLC仪,包括二极管阵列检测器(DAD)、色谱工作站(美国Agilent公司);XP-205型电子分析天平(瑞士Mettler-Toledo公司);KQ-250DV型超声波清洗仪(昆山市超声仪器有限公司,功率:250W,频率:40 kHz);M illi-QAdvantageA10型超纯水仪(美国M illipore公司)。
1.2 药品与试剂
维格列汀片(上海太昊生物科技医药有限公司,批号:150701、150702、150703,规格:50mg/片);维格列汀对照品(上海太昊生物科技医药有限公司,批号:131201,纯度:99.8%);杂质A对照品(批号:1448-001A2,纯度:97.6%)、杂质B对照品(批号:1607-005A3,纯度:95.7%)、杂质C对照品(批号:1424-093A3,纯度:99.6%)、杂质D对照品(批号:1497-003A7,纯度:98.1%)均购自中国食品药品检定研究院;乙腈、甲醇为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Xterra MSC18(50mm×4.6mm,3.5µm);流动相:[磷酸盐缓冲液-水-乙腈-甲醇(400∶600∶15∶15,V/V/V/V)](A)-[磷酸盐缓冲液-乙腈-甲醇(400∶450∶150,V/V/V)](B),梯度洗脱(0~1 m in,100%A;2~6 m in,100%→90%A;7~11 m in,90%→30%A;12~21 min,30%A;22min,30%→100%A;23~28m in,100% A);流速:1.2m L/min;检测波长:210 nm;柱温:35℃;进样量:10µL[6]。
2.2 溶液的制备
2.2.1 混合对照品溶液 分别精密称取杂质A对照品9.40mg、杂质B对照品11.32mg、杂质C对照品10.87 mg、杂质D对照品9.56mg,置于同一10m L量瓶中,加0.2%盐酸溶液-乙腈(90∶10,V/V)溶解并定容,摇匀,即得。
2.2.2 供试品溶液 取样品细粉适量,精密称定,置于50m L量瓶中,加0.2%盐酸溶液-乙腈(90∶10,V/V)适量,超声处理5min使溶解,加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45μm微孔滤膜滤过,即得(常温避光保存)。
2.2.3 阴性对照溶液 按样品的处方比例和制备工艺,取不含各待测杂质和主成分的空白辅料适量,按“2.2.2”项下方法制备阴性对照溶液。
2.2.4 系统适用性溶液 精密称取维格列汀对照品504.1 mg,置于50 m L量瓶中,加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,即得维格列汀对照品贮备液。分别精密称取杂质A、B、C、D对照品适量,置于同一100m L量瓶中,精密加入上述维格列汀对照品贮备液10m L,加0.2%盐酸溶液-乙腈(90∶10,V/V)溶解并稀释制成每1m L含维格列汀1mg和杂质A、B、C、D各10µg的系统适用性溶液。
2.3 系统适用性与专属性试验
精密量取“2.2”项下混合对照品溶液、供试品溶液、阴性对照溶液和系统适用性溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,在该色谱条件下,各成分均能达到基线分离,分离度均>1.5;理论板数以维格列汀峰计为8 700,保留时间为8.96 min。结果表明,其他成分对测定不干扰。
2.4 破坏性试验
2.4.1 酸破坏样品溶液 精密称取样品细粉适量(约相当于维格列汀50mg)(批号:150701),置于50m L量瓶中,加2mol/L盐酸溶液1m L,水浴(37℃)放置1 h,加2 mol/L氢氧化钠溶液调节pH为中性,加0.2%盐酸溶液-乙腈(90∶10,V/V)适量,超声处理5 m in使溶解,再加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
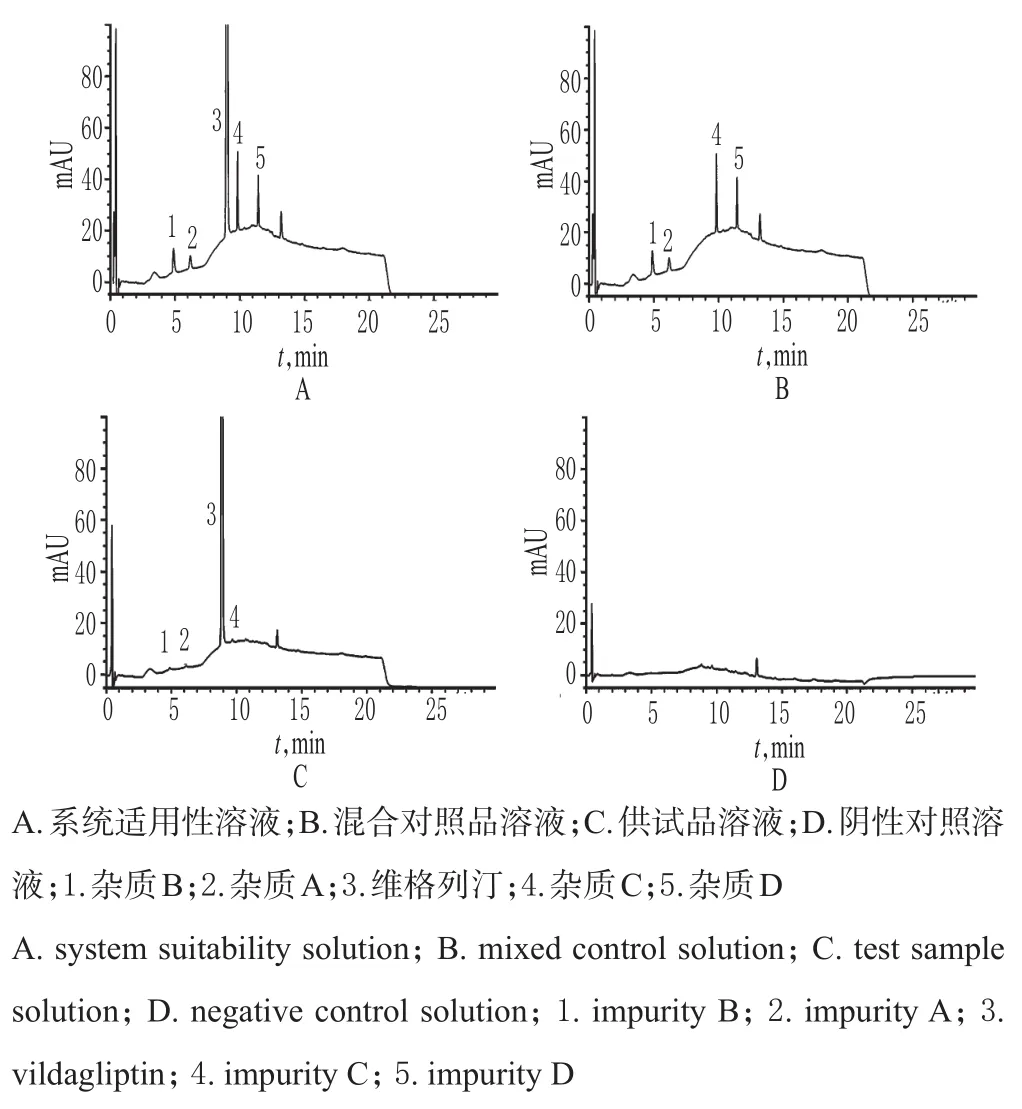
图1 系统适用性试验高效液相色谱图Fig 1 HPLC chromatogramsof system suitability tests
2.4.2 碱破坏样品溶液 精密称取样品细粉适量(约相当于维格列汀50mg)(批号:150701),置于50m L量瓶中,加0.02mol/L氢氧化钠溶液1m L,室温避光放置1 h,加0.02mol/L盐酸溶液调节pH为中性,加0.2%盐酸溶液-乙腈(90∶10,V/V)适量,超声处理5m in使溶解,再加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.4.3 氧化破坏样品溶液 精密称取样品细粉适量(约相当于维格列汀50mg)(批号:150701),置于50m L量瓶中,加30%过氧化氢溶液1m L,水浴(37℃)放置1 h,加0.2%盐酸溶液-乙腈(90∶10,V/V)适量,超声处理5 m in使溶解,再加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。
2.4.4 高温破坏样品溶液 精密称取样品细粉适量(约相当于维格列汀50mg)(批号:150701),置于50m L量瓶中,于105℃烘箱中避光放置3 h,加0.2%盐酸溶液-乙腈(90∶10,V/V)适量,超声处理5m in使溶解,再加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.4.5 光照破坏样品溶液 精密称取样品细粉适量(约相当于维格列汀50mg)(批号:150701),置于50m L量瓶中,于(12 000±500)lx光照强度下放置24 h,加0.2%盐酸溶液-乙腈(90∶10,V/V)适量,超声处理5m in使溶解,再加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得。
取上述各破坏样品溶液10µL,按“2.1”项下色谱条件进样测定,记录色谱,详见图2。由图2可知,样品在酸、碱、氧化、高温、光照破坏条件下均有一定程度的降解,但各降解产物之间及各降解产物与主峰之间均能达到良好分离(分离度>1.5)。
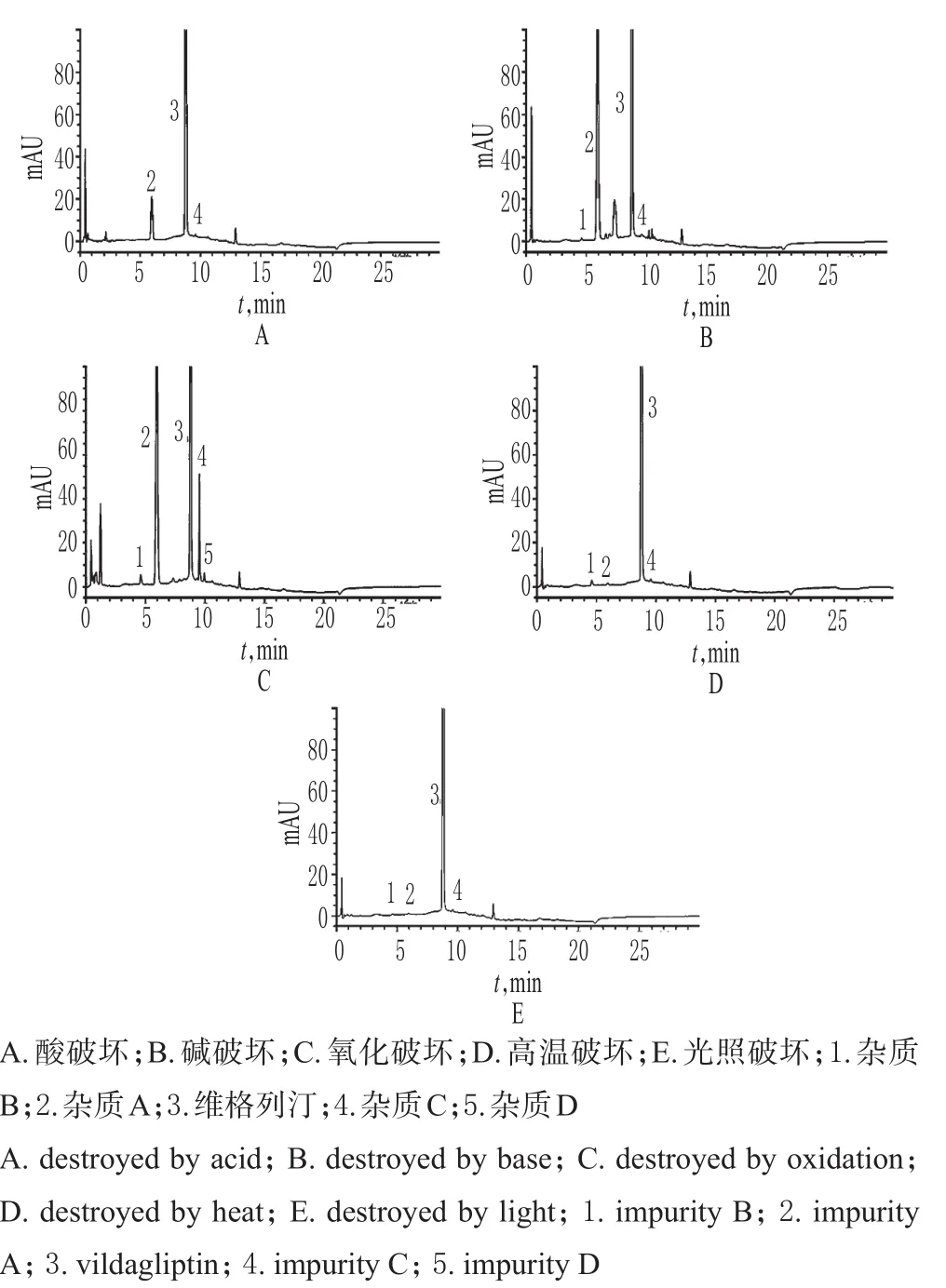
图2 破坏性试验高效液相色谱图Fig 2 HPLC chromatogramsofdestructive tests
2.5 线性关系考察
分别精密量取“2.2.1”项下混合对照品溶液1.0、2.5、5.0、7.5、10m L,分别置于50m L量瓶中,加0.2%盐酸溶液-乙腈(90∶10,V/V)定容,摇匀,经0.45μm微孔滤膜滤过,取续滤液,即得系列混合对照品溶液。取上述系列混合对照品溶液各适量,按“2.1”项下色谱条件进样测定,记录峰面积。以待测有关物质质量浓度(x,µg/m L)为横坐标、峰面积(y)为纵坐标进行线性回归,回归方程与线性范围见表1。
2.6 检测限考察
取“2.2.1”项下混合对照品溶液适量,倍比稀释,按“2.1”项下色谱条件进样测定,当信噪比为3∶1时,得检测限。结果,杂质A、B、C、D的检测限分别为4.18、2.68、 1.12、1.34µg/m L。

表1 回归方程与线性范围Tab 1 Regression equationsand linear ranges
2.7 精密度试验
精密量取“2.2.1”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,杂质A、B、C、D峰面积的RSD分别为0.98%、0.66%、0.71%、1.02%(n=6),表明仪器精密度良好。
2.8 稳定性试验
取“2.2.2”项下供试品溶液(批号:150701)适量,分别于室温下放置0、2、4、8、12、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,杂质A、B、C、D峰面积的RSD分别为2.89%、1.90%、1.66%、0.87%(n=6),表明供试品溶液在室温下放置24 h内基本稳定。
2.9 重复性试验
取样品(批号:150701)细粉适量(约相当于维格列汀50mg),共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积。结果,杂质A、B、C、D峰面积的RSD分别为0.56%、1.26%、0.77%、1.98%(n=6),表明本方法重复性良好。
2.10 加样回收率试验
取样品(批号:150701)细粉适量,共9份,分别置于50m L量瓶中,加入低、中、高质量的杂质A、B、C、D对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表2。
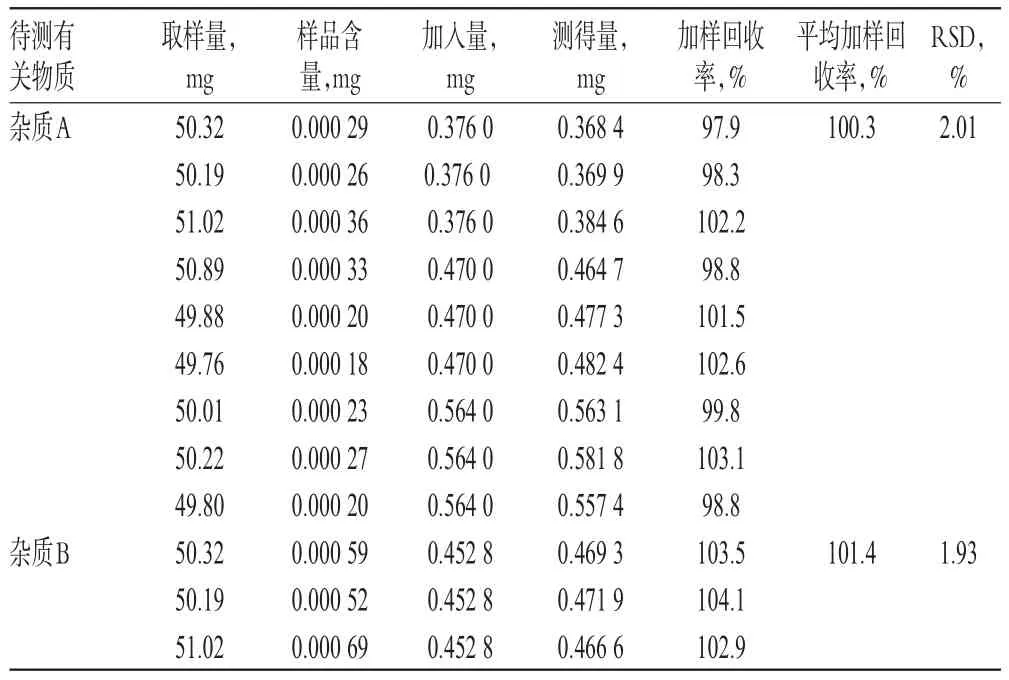
表2 加样回收率试验结果(n=9)Tab 2 Resultsof recovery test(n=9)

续表2Continued tab 2
2.11 样品有关物质测定
取3批样品细粉各适量(约相当于维格列汀50 mg),分别按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算各待测有关物质的含量[7-8],结果见表3(注:“-”为未检出)。

表3 样品有关物质测定结果(n=3,%)Tab 3 Determ ination of related substancesofsamples(n=3,%)
3 讨论
笔者采用DAD检测器对各待测有关物质进行全波长扫描,结果显示杂质A、B、C、D在200~400波长范围内均仅有末端吸收,辅料在210 nm处基本无吸收,不干扰各待测有关物质范围内的测定,因此最终选择210 nm作为本试验的检测波长。
综上所述,本方法操作简单、结果准确,可用于维格列汀片中有关物质的测定。
[1] IshiiM,Shibata R,Kondo K,etal.Vildagliptin stimulates endothelial cell network formation and ischemia-induced revascularization via an endothelial nitric-oxide synthase-dependentmechanism[J].JBiol Chem,2014,289(39):27235-27345.
[2] 吕晓川,王伟夫.二肽基肽酶Ⅳ抑制剂治疗2型糖尿病的临床研究进展[J].中国医药导报,2008,5(29):21-22.
[3] 刘甦苏,吴曦,周舒雅,等.3种小鼠品系的四氧嘧啶糖尿病模型建立及初步评价[J].药物分析杂志,2015,35(11):1958-1960.
[4] 樊新星,徐珽,卢静,等.抗糖尿病新药维格列汀[J].中国新药杂志,2008,17(14):1272-1274.
[5] 马廉举,文棘,胡煮,等.反相高效液相色谱法测定维格列汀[J].光谱实验室,2012,29(4):2555-2558.
[6] Shantikumar S,Satheeshkumar N,Srinivas R.Pharmacokinetic and protein binding profile of peptidom imetic DPP-4 inhibitor-Teneligliptin in rats using liquid chromatography-tandem mass spectrometry[J].JChromatogr B,2015(1002):194-200.
[7] 谢沐风.如何建立高效液相色谱法测定有关物质的方法[J].中国医药工业杂志,2007,38(1):45-47.
[8] 余振喜,庚莉菊,黄海伟,等.浅谈HPLC法测定有关物质时已知杂质的计算方法[J].中国药品标准,2010,11(4):278-279.
(编辑:刘 柳)
Determ ination of Related Substances in Vildagliptin Tabletsby HPLC
HUANG Shan1,ZHANG Mengze2(1.Henan Province Port Institute for Food Inspection and Testing,Zhengzhou 450003,China;2.Henan Provincial Institute for Food and Drug Control,Zhengzhou 450003,China)
OBJECTIVE:To establish amethod for the determination of related substances in Vildagliptin tablets.METHODS:HPLC method was adopted.The determination was performed on Xterra MSC18column w ith mobile phase consisted of[phosphate buffer-water-acetonitrie-methanol(400∶600∶15∶15,V/V/V/V)]-[phosphate buffer-acetonitrile-methanol(400∶450∶150,V/V/V)](gradient elution)at the flow rate of 1.2m L/min.The detection wavelength was set at 210 nm and column temperature was 35℃. The sample size was 10µL.RESULTS:The linear ranges were 18.80-188.0µg/m L for impurity A(r=0.999 5),22.64-226.4µg/ m L for impurity B(r=0.999 6),21.74-217.4µg/m L for impurity C(r=0.999 7),19.12-191.2µg/m L for impurity D(r=0.999 8).The lim its of detection were 4.18,2.68,1.12,1.34µg/m L,respectively;RSDs of precision,stability and reproducibility tests were lower than 3%;the recoveries of impurity A,B,C and D were 97.9%-103.1%(RSD=2.01%,n=9),98.8%-104.1%(RSD=1.93%,n=9),98.0%-103.6%(RSD=1.81%,n=9),100.8%-104.1%(RSD=0.98%,n=9),respectively.CONCLUSIONS:Themethod is simple and accurate,and can be used for the determ ination of related substances in Vildagliptin tablets.
Vildagliptin tablets;Related substances;HPLC
R927
A
1001-0408(2017)15-2138-04
2016-06-30
2017-01-12)
*主管药师,硕士。研究方向:食品、药品分析及质量标准。电话:0371-66081055。E-mail:hs13695541@163.com
DOI 10.6039/j.issn.1001-0408.2017.15.34
猜你喜欢
中国兽药杂志(2022年6期)2022-07-04
中国药学药品知识仓库(2022年13期)2022-07-03
中国典型病例大全(2022年7期)2022-04-22
药品评价(2021年17期)2021-11-06
药品评价(2020年24期)2020-12-17
儿童文学选刊(2019年7期)2019-09-10
天津药学(2019年4期)2019-09-10
人生十六七(2014年12期)2014-11-27
药品评价(2012年34期)2012-12-08
药学研究(2012年2期)2012-10-25
