
基于分子对接虚拟筛选MEK1中药抑制活性成分
2017-06-19 18:14黎永良杜志云郑杰
中国中药杂志 2017年10期
关键词:分子对接
黎永良+杜志云+郑杰

[摘要] 有丝分裂原活化蛋白激酶激酶1(mitogen-activated protein kinase kinase,MEK1)的过度磷酸化是黑色素瘤成因之一,基于该靶点的中药成分虚拟筛选有望发现中药在治疗黑色素瘤潜在应用价值。采用MEK1晶体构象构建口袋模型和Flex Search模型,模型可很好重现晶体结构,对接配体构象与晶体结构中原配体构象similarity评分为0.784;对接评分与已有MEK1抑制剂活性数据pIC50呈线性关系,R2=0.937,进一步表明虚拟筛选模型可靠。以口袋模型对Lipinski五规则初筛的中药成分库进行分子对接,根据Flex Search模型精筛,最终得到50个总分高于7.0的化合物,该研究给出总分高于阳性对照的前10个中药成分,有望进一步用于MEK1和黑色素瘤抑制的研究。
[关键词] MEK1抑制剂; 中药成分; 分子对接; 虚拟筛选
[Abstract] The hyperphosphorylation of MEK1(mitogen-activated protein kinase kinase 1) is one of the causesfor melanoma. It is important to discover a potential medicine for melanoma through virtual screening of chemical composition of Chinese material medica on MEK1. In this study, a docking pocket model and Flex Search model were built by using a MEK1 crystal structure, the similarity between connector conformation and gametophyte conformation in the crystal structure was 0.784.There was a linear relationship between total score and pIC50on MEK1 inhibitors, with R2=0.937, which further indicated the reliability of the virtual screening model.The search library of traditional Chinese medicine database on the Lipinski′s rule was docked with the pocket model, and then 50 compounds with a totalscore of more than 7.0 were obtained by the Flex model. In this paper, top 10 active ingredients in screening results showed atotal score of more than that of positive medicines. It is expected to further research the inhibition of MEK1 and melanoma.
[Key words] MEK1 inhibitor; chemical composition of Chinese material medica; molecular docking; virtual screening
蛋白激酶是一種高度多样性的酶,不同活性状态的激酶可引起细胞的不同反应[1]。Raf-MEK1/2-ERK1/2三级激酶级联反应主要影响细胞的生长、增殖和分化[2-3],Raf的突变将引起下游因子MEK1/2的过度磷酸化,导致有丝分裂过度进行。90%黑色素瘤在蛋白组学上表现为MAPK信号通路的过度磷酸化,其中50%为c-Raf和b-Raf的突变[4]。MEK1/2的抑制剂主要有两大类:Raf-MEK1/2结合抑制剂以及被多数应用的MEK1/2异构抑制剂,包括Trametinib,PD098059,UO126,PD183452都是很好的异构抑制剂,尤其Trametinib对c-Raf激活的MEK1/2的IC50分为0.92,1.8 nmol·L-1,被FDA认可为黑色素瘤治疗药,然而PD098059,UO126,PD183452在临床研究由于吸收、代谢原因没有被采用[5-6]。
天然产物是生物在适应生存环境下产生的次级产物,具有维持生理和自身防御的功能,具有结构多样性、新颖性、复杂性和副作用小的特点[7],在抗感染、抗病毒、抗肿瘤、免疫和中枢神经系统药物方面具有优势,尤其临床上应用的喜树碱、紫杉醇、鬼臼毒素等抗肿瘤药物均来自天然产物。中药作为天然药物来源有3万多种,常用的有300~400种,其含有的化学成分有可能用于开发抗肿瘤药物,如甘草中含有的光甘草定已有报道抑制MEK1具有抗肿瘤作用[8]。
计算机辅助药物设计是常用的药物研发方法,主要包括虚拟筛选和药效团设计[9]。本研究采用被解析的MEK1晶体结构建立抑制成分筛选模型,并利用该模型筛选中药成分数据库中MEK1抑制活性成分,对中药在抗肿瘤尤其是抗黑色素瘤活性研究和开发上具有导向意义。
1 材料与方法
1.1 MEK1抑制剂虚拟筛选模型的构建
1.1.1 MEK1结合口袋的构建 在Protein Data Bank数据库中搜寻具有抑制剂的MEK1共晶结构晶体(PDB code:4LMN),该蛋白共晶结构为MEK1与别构抑制剂GDC0973的复合物,GDC0973与MEK1催化区域附近的Asp190和Asn195形成氢键并结合于MEK1的催化区域[6]。利用Sybyl-X软件的Biopolymer模块提取GDC0973并删除其余配体,为蛋白加氢及修复末端残基,设置阈值(Threshold)和膨胀系数(Bloat)分别为0.70,1,利用Docking模块以配体作为模板形成抑制剂结合口袋[10]。
1.1.2 MEK1的Flex Search模型构建 利用Site ID识别抑制剂结合口袋并形成范德华力表面作为限制范围,指定MEK1与抑制剂形成氢键的氨基酸残基Lys97,Asp208,Phe209,Ser212,Ser 212[6,11]3内的残基作为氢键的供体和受体,选择指向于结合口袋的氢键作为限制条件,所有条件的形成参数均为默认参数。以FlexSearch作为进一步的筛选方法[12],当同时满足在范德华力的限制表面范围内,且筛选对象同时与受体形成2个或2个以上氢键时被确认为候选分子。
1.1.3 MEK1抑制剂虚拟筛选模型的验证 利用Sybyl-X中Surflex-Dock模块,将GDC0973作为配体与目标模型进行对接,起始构象个数为6个,原配体构象作为参考模板,考察相似度分数大小[8];同时选取Trametinib,Cobimetinib,RO49877655,Binimetinib,Selumetinib这5个已知的MEK1抑制剂对模型进行对接评分和Flex Search筛选验证[13]。
1.2 中药成分分子库优化和虚拟筛选
1.2.1 中药成分分子库获取与优化 中药成分数据库来自ZINC,选用TCM Database @ Taiwan数据库[14],该数据库是中药化学成分库,含有4.9万个源于中药成分。对TCM Database @ Taiwan分子库中分子构象加载Gasteiger-Hückel电荷,采用Tirpos力场Minimize程序进行能量最小化,以最大次数3 000,0.001 kcal·mol-1作为收敛标准的Powel能量梯度法进行优化,得到稳定构象[10]。
1.2.2 中药分子库的虚拟筛选 中药分子库虚拟筛选由3个步骤构成:首先,经过能量优化的中药分子采用Lipinski五规则进行初步筛选;其次针对Lipinski五规则筛选的化合物用Surflex-Dock分子对接的方法评分,获得配体-受体结合最优构象;最后利用Flex Search进行进一步构象筛选,最终获得候选化合物。
2 结果与分析
2.1 MEK1抑制剂虚拟筛选模型
以MEK1晶体结构(PDB:4LMN)的配体GDC0973作基础,设置阈值和膨胀系数分别为0.70,1,选用“配体”作为参考模板生成结合口袋。经PyMOL软件[15]制作见图1,显示口袋与配体GDC0973之间形成氢键的残基是Lys97,Asp190,Asn195和Ser212,处于MEK1的α-C端螺旋附近,该位置为MEK1被Raf磷酸化和催化ERK磷酸化的活性区域[6],配体与残基Asp208,Met219所在区域形成疏水作用。结合口袋完全处于MEK1蛋白肽链的结构以内,使Surflex-Dock程序在对接过程中配体不溢出MEK1蛋白的结构,初步认为该模型可用于分子对接的虚拟筛选。
2.2 MEK1的Flex Search 模型
通过SiteID识别抑制剂结合口袋并建立范德华表面,并以MEK1抑制剂作用残基为中心,选择3范围内的残基构建MEK1的Flex Search模型。
MEK1关键氨基酸3范围内残基共35个,其中氢键取向正确的仅15个,作为氢键受体的残基包括Asn78,Gly80,Asp190,Asn195,Asp208,Phe209,Ser212,Ile216,Met219,Asn221,Phe223,Val224,Arg234,另外Asn78,Lys97,Lys192,Asp208,Ser212和Arg234作为氢键供体结合配体,这2类氨基酸残基主要分布在Lys97附近,具有氢键供体的配体可能更加具有结合优势。特别地,Lys97,Asp208,Ser212在抑制口袋中主要作为氢键供体与抑制剂形成氢键[4,16],而3范围内的其他氨基酸残基则主要作为氢键的受体与配体形成氢键。
2.3 MEK1抑制剂虚拟筛选模型的可靠性验证
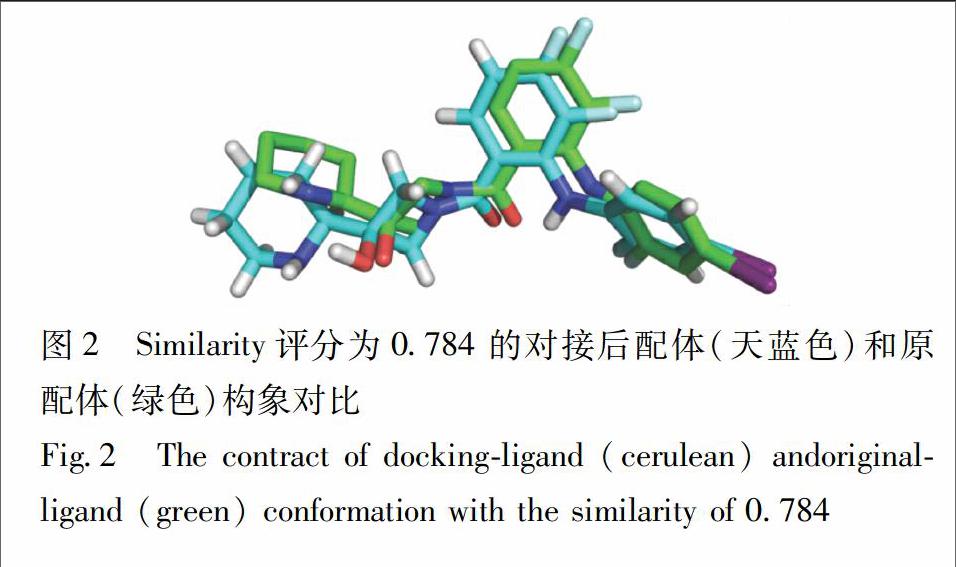
MEK1模型构建是基于半柔性对接的设计,认为配体是可以变化的,这种对接方式适用于大分子和小分子的对接,因此有必要针对模型进行对接参数的验证。利用Sybyl-X的Surflex-Dock模块将提取的GDC0973作为配体与目标模型对接,对接构象与原有配体对比进行相似度评分,认为相似度分数大于0.7即可用于分子对接筛选[8],对接构象与原有配体构象对比叠合较好,其相似度分数为0.784,说明该对接模型能重现4LMN晶体结构,可用于后续分子对接虚拟筛选,见图2。
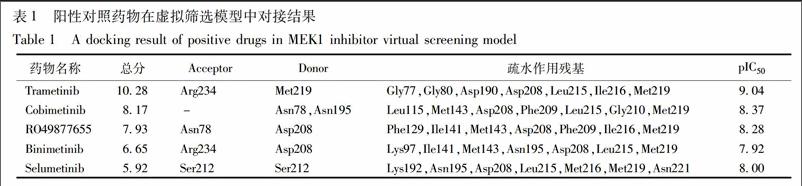
将5种已知的MEK1抑制剂通过Surflex-dock模块进行分子对接,对模型的可靠性进一步验证。通过对接评分总分(Total Score),Flex Search得到的氢键供体和受体氨基酸残基进行考察。5种已知MEK1抑制剂在该模型中的对接评分、氢键受体氨基酸残基(Acceptor)、氢键供体氨基酸残基(Donor)和对应的pIC50见表1。结果表明5个阳性药物的对接评分在5~10分,对接评分与pIC50之间具有良好的线性关系(R2=0.937);在Flex Search中显示他们可以与Asn78,Asn195,Asp208,Ser212,Met219,Arg234氨基酸残基形成氢键,以LigPlot[18]分析阳性药与MEK1疏水作用(仅给出较强的7个残基)均包括Asp208和Met219及附近残基,已知MEK1抑制剂的分子对接試验结果进一步说明该虚拟筛选模型是可靠的。
2.4 中药化学成分虚拟筛选
对TCM Database @ Taiwan数据库的4.9万个化合物以Lipinski五规则进行初步筛选,得到18 752个待选化合物,下一步采用Surflex-Dock进行对接筛选,得到2 448个总分大于7.0的化合物,随后进一步采用Flex Search精筛,共得到50个化合物。本研究按照总分为第一、QFIT为第二的排序条件进行排序并列出前10个最优化合物,分析氢键作用和疏水作用残基(给出作用较强10个残基)见表2。前10个化合物总分均大于10.28,与MEK1可形成5个氢键以上,可能具有比阳性药物更好的作用效果,其中Gly77,Asn78,Gly80,Lys97,Val127,His188,Lys192,Arg189,Asp190,Asn195,Asp208,Phe209,Val211,Ser212,Met219,Asn221,Ser222,Tyr229,Arg234可分别与前10化合物形成氢键,疏水分析显示前10化合物均与关键疏水残基形成疏水作用,说明这些化合物可以较好地与MEK1的结合口袋作用。根据化合物官能团进行归类,大部分为含有羟基和氨基的化合物。
其中ZINC85595772为最优成分,总分、作用力评分(Polar)和QFIT评分都较好,与MEK1晶体模型形成8个氢键,特别地,针对该成分进行对接模式的分析见图3。图3中A显示ZINC85595772结合于MEK1的抑制口袋中,与口袋周围Asn78,Asn195,Asp208,Ser212,Try229形成氢键,且可以与同一个氨基酸残基形成多个氢键,对比图3中B配体GDC0973与MEK1结合位点为Lys97,Asp190,Asn195,Ser212。其中Ser212是MEK1磷酸化功能的最关键氨基酸残基[16],作为氢键供体与ZINC85595772羟基氧和GDC0973的氟原子相互作用;分析配体在MEK1的疏水作用,图3中C可观察ZINC85595772主要与Asn78,Lys97,Asp190,Asn195,Asp208,Ser212,Met219,Tyr229,Asn221,Met230形成较强疏水作用,与图3中D的GDC0973配体对比具有相似的疏水作用,尤其是比较关键的Asp208,Met219残基。说明ZINC85595722具有与GDC0973相似的结合模式,可能是潜在的MEK1抑制剂,根据对接残基和总分推测其他化合物也具有潜在的MEK1抑制活性。
3 讨论
中药含有的化合物种类较多且活性大部分不明确,因此针对抑制位点的中药活性成分筛选可期望发现具有较好活性、副作用低的中药来源化合物。90%的黑色素瘤在MAPK信号通路上有高度的活性[3,6],针对抑制MEK1/2高度磷酸化的中药活性成分筛选,可为治疗黑色素瘤的药物研发提供新的物质基础和参考。本研究采用已有的抑制剂-MEK1共晶结构,构建MEK1抑制活性成分Surflex-Dock和Flex Search筛选模型,利用该模型对中药成分进行筛选并分析其作用模式。
MEK1模型构建方面,序号为4LMN的MEK1晶体结构Rfree=0.233,Rwork=0.191,分辨率2.8,氨基酸序列与其他晶体相比较为完整,误差较少,满足虚拟筛选模型构建要求。本研究构建2种模型用于对接和精筛:首先提取配体于MEK1磷酸化活性关键位点[6]构建Surflex-Dock对接模型,确保筛选位点的准确性;建立Flex Search模型则要求配体结合于抑制口袋并与位点附近残基形成至少2个氢键,使化合物与MEK1的复合物处于稳定状态,进一步精筛对接结果。Flex Search模型显示MEK1抑制剂结合位点对配体的要求主要以氢键的供体为主,推测多酚类、氨基类的结构可能会有比较好的抑制活性。
模型可靠性验证方面,先通过提取的配体与模型对接,对接构象与原构象相似度分数为0.784,说明口袋模型有较好的构象重现性;其次选择5个已知MEK1抑制剂与口袋模型进行分子对接和Flex Search搜寻验证模型可靠性,发现5个MEK1抑制剂对接分数与pIC50线性关系良好(R2=0.937),Flex Search搜寻结果显示这些MEK1抑制剂与文献报道的关键残基有氢键和疏水作用,说明对接模型对阳性药活性预测较好。总的而言,说明该模型有较好的预测能力。
中药成分筛选结果方面,总分、polar、QFIT评分越高,理论上作用效果越好,本研究最终筛选得到50个活性成分并列出总分前10化合物总结其结构类型,选择打分最高的ZINC85595772进行结合模式分析。评分前10化合物主要含有羟基和氨基,总分均比阳性对照化合物高,与结合位点附近残基形成至少5个氢键,这些残基与已有抑制剂的氢键作用均在文献中报道[6,11,17],属于5种阳性药物与MEK1氢键作用残基3范围内,化合物与PDB晶体结构中Asp208,Met219及附近残基均有疏水残基作用,说明它们的结合模式与目前所应用的MEK1抑制剂相似。特别地,ZINC85595772和GDC0973均结合在MEK1磷酸化位点Ser212附近,从氢键结合和位阻上抑制Ser212的磷酸化,推测对MEK1有抑制作用。前10化合物依据总分以及作用残基认为具有潜在的MEK1抑制活性,总分高于模型验证的线性关系上限,有待进行MEK1的IC50实验。
4 结论
本研究以MEK1晶体及其配体构象为模板构建口袋模型和Flex Search模型并证实模型的可靠性,通过Lipinski五规则初步筛选、Surflex-Dock对接和Flex Search搜寻对4.9万个中药化合物进行虚拟筛选,最终得到50个对接分数7.0分以上的化合物,评分前10的成分有望进行下一步MEK1和黑色素瘤抑制活性研究。Surflex-Dock对接和Flex Search模型显示,含有羟基、氨基的中药成分是比较理想的MEK1抑制剂,该类成分含量较大的中药有期望抑制Raf-MEK1/2-ERK1/2信号通路和黑色素瘤,这对中药研究具有很好参考价值。
[參考文献]
[1] Jonathan M, Elkins S K. The structure of the full-length tetrameric PKA regulatory RIIβ complex reveals the mechanism of allosteric PKA activation[J]. Sci Signaling, 2012(5): pe21.
[2] Wellbrock C, Arozarena I. The complexity of the ERK/MAP-kinase pathway and the treatment of melanoma skin cancer[J]. Front Cell Dev Biol, 2016(4): 33.
[3] Roskoski R. ERK1/2 MAP kinases: structure, function, and regulation[J]. Pharmacol Res, 2012, 66(2): 105.
[4] Luke J J, Ott P A, Sharpiro G I. The biology and clinical development of MEK inhibitors for cancer[J]. Drugs, 2014, 74(18): 2111.
[5] Yamaguchi T, Kakefuda R, Tajima N, et al. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo[J]. Int J Oncol, 2011, 39(1): 23.
[6] Shang J, Lu S, Jiang Y, et al. Allosteric modulators of MEK1: drug design and discovery[J]. Chem Biol Drug Des, 2016, 88(4): 485.
[7] Sheng H, Sun H. Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi-target approach to prevention and treatment of metabolic and vascular diseases[J]. Nat Prod Rep, 2011, 28(3): 543.
[8] Wang Z, Luo S, Wan Z, et al. Glabridin arrests cell cycle and inhibits proliferation of hepatocellular carcinoma by suppressing braf/MEK signaling pathway[J]. Tumor Biol, 2015, 37(5): 5837.
[9] 乔连生,张燕玲. 计算机辅助药物设计在天然产物多靶点药物研发中的应用[J]. 中国中药杂志, 2014,39(11):1951.
[10] 杜志云,汤志恺,卢宇靖,等. 姜黄素类似物与酪氨酸酶相互作用的分子对接研究及应用[J]. 计算机与应用化学, 2011, 28(5):531.
[11] Dong Q, Dougan D R, Gong X, et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer[J]. Bioorg Med Chem Lett, 2011, 21(5): 1315.
[12] Nicholas Rhodes, Clark D E, Willett A P. Similarity searching in databases of flexible 3D structures using autocorrelation vectors derived from smoothed bounded distance matrices[J]. J Chem Inf Model, 2006, 46(2): 4.
[13] 周月, 张兵, 林志健, 等. 基于分子对接技术虚拟筛选菊苣与肠道CNT2结合的化学成分研究[J]. 中国中药杂志, 2016, 41(21): 3962.
[14] Chen C Y-C. TCM Database@Taiwan: The World′s largest traditional Chinese medicine database for drug screening in silico[J]. PLoS ONE, 2011, 6(1): e15939.
[15] Schrodinger LLC(2010). The PyMOL molecular graphics system, version 1.3r1[EB/OL]. http://www.pymol.org.
[16] Hatzivassiliou G, Haling J R, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers[J]. Nature, 2013, 501(7466): 232.
[17] Isshiki Y, Kohchi Y, Iikura H, et al. Design and synthesis of novel allosteric MEK inhibitor CH4987655 as an orally available anticancer agent[J]. Bioorg Med Chem Lett, 2011, 21(6): 1795.
[18] Wallace A C, Laskowski R A, Thornton J M.LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions[J].Protein Eng, 1995, 8(2):127.
[責任编辑 张宁宁]
猜你喜欢
