
Schwartz-Jampel综合征1A型一例临床特点分析
2017-06-19 19:25:50周海涛任向阳马聪敏滕军放
中国全科医学 2017年18期
黄 超,周海涛,任向阳,马聪敏,滕军放
·临床诊疗提示·
Schwartz-Jampel综合征1A型一例临床特点分析
黄 超1,周海涛1,任向阳1,马聪敏1,滕军放2*
目的 分析1例Schwartz-Jampel综合征1A型患儿的临床特点。方法 2016-06-15至2016-08-15,回顾性分析1例就诊于郑州大学附属洛阳中心医院的Schwartz-Jampel综合征1A型患儿的临床资料,总结其主要临床表现以及肌肉病理染色、基因检测结果。结果 主要临床表现为面部表情固定,睑裂狭小,睁眼费力,小口,张口费力,小下颌,步态略僵硬;肌酸肌酶水平轻度升高;肌电图提示肌源性损害,静止时有大量肌强直电位发放。肌肉病理染色结果显示肌内衣结缔组织轻度增生,肌纤维直径变异轻度加大,少数散在及成组分布的陈旧坏死肌纤维伴随炎性细胞浸润及嗜碱性肌纤维伴随核肥大,个别分裂肌纤维,少数肌纤维核内移。基因检测结果显示患儿HSPG2基因呈复合杂合突变:10号外显子c.10736T>G(p.Ile3579Ser),73号外显子c.9963C>A(p.Cys3321Ter),77号外显子c.1208G>A(p.Cys403Tyr)。结论 结合患儿的临床表现、病理改变特点及基因结果,Schwartz-Jampel综合征1A型诊断明确。特殊的面部改变是本病最显著的临床特点,肌肉病理呈肌营养不良样改变。
骨软骨发育不良;肌强直;HSPG2基因
黄超,周海涛,任向阳,等.Schwartz-Jampel综合征1A型一例临床特点分析[J].中国全科医学,2017,20(18):2271-2273.[www.chinagp.net]
HUANG C,ZHOU H T,REN X Y,et al.Clinical features of one case of Schwartz-Jampel syndrome type 1A[J].Chinese General Practice,2017,20(18):2271-2273.
Schwartz-Jampel综合征(SJS)是一种罕见的常染色体隐性遗传病,1962年由SCHWARTZ和JAMPEL首次报道,其主要临床特点包括特殊的面部特征:肌强直诱发的眼睑痉挛,固定的面部表情,撅嘴,小口,多行长睫毛;肢体僵硬和无力导致患者呈下蹲姿势和鸭步步态;骨骼异常:身材矮小,鸡胸,脊柱后侧突和关节挛缩[1]。GIEDIOND等根据患者临床表现和骨骼X线特点将SJS分为1A、1B、2型,SJS-1A型与SJS-1B型临床表现类似,但SJS-1A型一般于婴儿期以后起病,存在轻中度骨发育不良,SJS-1B型于出生后即刻发病,伴有严重的骨发育不良[2]。目前研究发现,SJS-1型与HSPG2基因突变有关;SJS-2型又称为Stuve-Wiedemann综合征,病死率较高,与LIFR基因突变有关[2-3]。我国目前报道SJS仅6例,基因确诊1例[3-5]。本文现报道经基因确诊的1例SJS-1A型患儿,并分析其临床特点及肌肉病理改变,旨在提高临床医生对该病的认识。
1 资料与方法
1.1 临床资料 患儿男,4岁,因“面部表情异常3年”于2016-06-15就诊于郑州大学附属洛阳中心医院。患儿足月顺产,运动发育里程碑正常。患儿1岁起出现面部肌肉发僵,面部表情减少,后逐渐发展为眼睑痉挛,睑裂狭小,睁眼费力,口部紧缩,张口费力,四肢活动尚可,无吞咽困难、饮水呛咳,无骨骼畸形。1岁时,曾因“急性扁桃体炎”于当地医院就诊,当时查肌酸肌酶(CK)1 111.5 U/L(参考范围25.0~200.0 U/L)。父母均表型正常,非近亲结婚,家族中无类似疾病。体格检查:身高95 cm,体质量15 kg。意识清楚,吐词略欠清,面部肌肉僵硬,睑裂狭小,双侧眼球各向活动充分,口唇发紧,张口费力,小下颌,四肢深浅感觉正常,四肢肌力5级,四肢肌张力轻度升高,步态略僵硬,四肢腱反射对称减低,未引出病理征,共济运动正常,无脑膜刺激征。辅助检查:CK 355.0 U/L。肌电图提示肌源性损害,静止时有大量肌强直电位发放。感觉及运动神经传导速度均正常。
经医院伦理委员会批准,患儿家属同意并签署知情同意书后,取患儿左侧肱二头肌活检组织进行病理检查。组织标本于异戊烷预冷后,在液氮中冰冻固定,冷冻切片,进行组织学、酶组织化学、免疫组织化学染色,包括苏木素-伊红(HE)、改良Gomori三色(MGT)、油红“O”脂肪(ORO)、高碘酸Schiff反应(PAS)、三磷腺苷酶(ATP酶),还原型辅酶Ⅰ四氮唑还原酶(NADH-TR)、琥珀酸脱氢酶(SDH)、肌营养不良素-N/C/R(Dystrophin-N、-C、-R)、α/β/γ-肌聚糖蛋白(α/β/γ-Sarcoglycan)、奇异不良素(Dysferlin)染色。抽取患儿及其父母外周血各2 ml送基因公司进行基因检测,将患儿外周血进行遗传性肌肉病相关基因测序,其父母外周血行家系验证。最终结合患儿的临床表现、病理改变特点及基因结果,诊断为SJS-1A型。
1.2 研究方法 2016-06-15至2016-08-15,回顾患儿的临床资料,总结其主要临床表现以及肌肉病理染色、基因检测结果。
本研究价值:
Schwartz-Jampel综合征(SJS)是一种罕见的常染色体隐性遗传疾病,1962年由SCHWARTZ和JAMPEL首次报道,目前我国报道的SJS仅6例,基因确诊1例。本文报道了1例罕见SJS病例,临床资料详细,且有病理及基因检测为支持依据,有助于提高临床医生对该病的认识。
2 结果
2.1 肌肉病理染色结果 HE染色示肌束衣边界清,肌内衣结缔组织轻度增生,肌间质未见炎性细胞浸润,肌纤维直径变异轻度加大,少数散在及成组分布的陈旧坏死肌纤维伴随炎性细胞浸润及嗜碱性肌纤维伴随核肥大,个别分裂肌纤维,少数肌纤维核内移(见图1,本文图1彩图见本刊官网www.chinagp.net电子期刊相应文章)。MGT染色未见破碎红纤维(RRF)。ORO染色未见肌纤维内脂肪滴增多。PAS染色未见肌纤维内糖原增多或缺乏。ATP酶染色显示两型纤维呈棋盘格分布,未见同型纤维群组化现象。NADH-TR染色显示肌纤维内氧化酶活性分布均匀。SDH染色未见血管壁强染现象(SSV)及破碎蓝纤维(RBF)。Dystrophin-N、-C、-R,α/β/γ-Sarcoglycan,Dysferlin染色均可见肌纤维膜阳性表达。
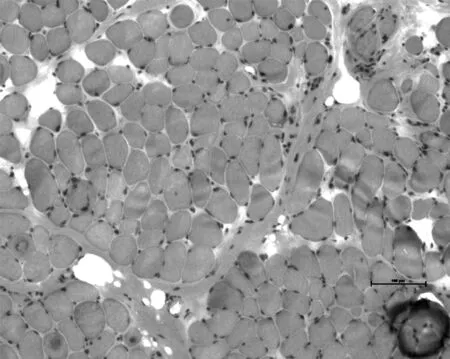
图1 患儿肌肉病理染色结果(HE染色,×100)
Figure 1 Pathological findings of the child patient′s biopsy specimens from the left biceps brachii muscle
2.2 基因检测结果 患儿HSPG2基因呈复合杂合突变:10号外显子c.10736T>G(p.Ile3579Ser),为错义突变,来自父亲;73号外显子c.9963C>A(p.Cys3321Ter),为无义突变,来自母亲;77号外显子c.1208G>A(p.Cys403Tyr),为错义突变,来自父亲。
3 讨论
本例患儿早期运动发育里程碑正常,于幼儿期起病,主要临床表现为面部表情固定,睑裂狭小,睁眼费力,小口,张口费力,小下颌,步态略僵硬;CK水平轻度升高;肌电图提示肌源性损害,静止时有大量肌强直电位发放。肌肉病理染色结果提示肌营养不良样病理改变特点。基因检测结果提示HSPG2基因复合杂合突变,来源于其父母,而其父母均正常,故遗传方式考虑为常染色体隐性遗传。结合患儿的发病年龄、临床表现、肌电图、肌肉病理染色及基因检测结果,目前诊断明确为SJS-1A型。
SJS-1A型的临床表现具有特异性,身材矮小者占90%,肌强直者占85%,撅嘴及小口者占80%,肌肉肥大者占70%,固定的面部表情者占55%,骨骼异常者占45%,CK水平升高者占45%,髋关节发育不良者占40%,眼睑痉挛及睑裂狭小者占32.5%[1,6]。
骨骼异常包括脊柱畸形、关节挛缩、骨质疏松、骨龄延迟等。有研究显示,SJS-1A型少见的临床特点包括高嗓音、双侧腕管综合征和恶性高热[6]。本例患儿具有典型的面部表现特征,但尚无骨骼异常的表现,可能与病程相对较短有关。
本研究SJS-1A型患儿的病理改变为肌内衣轻度增生,个别肌纤维肥大及萎缩,分散成组的肌纤维坏死及再生,符合肌营养不良样病理改变特点,与相关文献报道一致[7]。亦有研究报道该病可以在肌肉改变的基础上合并神经源性改变[5],故该病不同于骨骼肌缺乏明显病理改变的非肌营养不良性肌强直,行肌肉活检可予以鉴别诊断。
SJS目前明确的致病基因为HSPG2基因,其位于1号染色体P34-36,是编码基底膜蛋白多糖的基因[8]。基底膜蛋白多糖是细胞膜的重要组成部分,该蛋白的结构或功能缺陷可引起软骨发育不良和异常的神经肌肉活动[8-9]。该患儿基因检测结果提示HSPG2基因复合杂合突变:c.10736T>G(p.Ile3579Ser),c.9963C>A(p.Cys3321Ter),c.1208G>A(p.Cys403Tyr),上述突变均不属于多态性变化,属尚未见报道的新发突变。
综上所述,SJS是一种罕见的常染色体隐性遗传病,本例为第2例基因诊断明确的中国患儿。本文通过分析该病的临床及病理特点,旨在提高临床医生对本病的认识,虽该病尚无特效的治疗方法,但早期识别、明确诊断,应用离子通道阻滞剂、物理康复治疗可减轻症状,减缓疾病进程,后期配合面部整形及骨科矫形手术治疗,可改善患儿生活质量[10]。
作者贡献:黄超进行文章的构思与设计、数据收集、结果分析与解释、撰写论文;周海涛进行研究的实施与可行性分析;任向阳进行论文修订;滕军放负责文章的质量控制及审校;马聪敏对文章整体负责、监督管理。
本文无利益冲突。
[1]ABURAHMA S K,AL-KHATEEB T,ALREFAI A,et al.Botulinum toxin A injections for the treament of Schwartz-Jampel syndrome:a case series[J].J Child Neurol,2008,24(1):5-6.DOI:10.1177/0883073808320621.
[2]MALLINENI S K,YIU C K,KING N M,et al.Schwartz-Jampel syndrome:a review of the literature and case report[J].Spec Care Dentist,2012,32(3):105-111.DOI:10.1111/j.1754-4505.2012.00249.x.
[3]代丽芳,方方,黄昱,等.儿童Schwartz-Jampel综合征一例并文献复习[J].中华儿科杂志,2015,53(11):855-858.DOI:10.3760/cma.j.issn.0578-1310.2015.11.012. DAI L F,FANG F,HUANG Y,et al.Clinical and genetic features of Schwartz-Jampel syndrome in a Chinese child:case report and literature review[J].China J Pediatr, 2015,53(11):855-858.DOI:10.3760/cma.j.issn.0578-1310.2015.11.012.
[4]张坤,吴沪生,吕俊兰.Schwartz-Jampel综合征四例分析[J].中华儿科杂志,2012,50(3):231-234. ZHANG K,WU H S,LYU J L.Clinical analysis of four patients with Schwartz-Jampel syndrome[J].China J Pediatr,2012,50(3):231-234.
[5]郑日亮,栾兴华,陈彬,等.Schwartz-Jampel综合征一例[J].中华神经科杂志,2008,41(3):215.DOI:10.3321/j.issn:1006-7876.2008.03.024. ZHENG R L,LUAN X H,CHEN B,et al.Schwartz-Jampel syndrome:a case report[J].China J Neurol,2008,41(3):215.DOI:10.3321/j.issn:1006-7876.2008.03.024.
[6]BASIRI K,FATEHI F,KATIRJI B.The Schwartz-Jampel syndrome:case report and review of literature[J].Adv Biomed Res,2015,4:163.DOI:10.4103/2277-9175.162538.
[7]IWATA S,ITO M,NAKATA T,et al.A missense mutation in domain Ⅲ in HSPG2 in Schwartz-Jampel syndrome compromises secretion of perlecan into the extracellular space[J].Neuromuscul Disord,2015,25(8):667-671.DOI:10.1016/j.nmd.2015.05.002.
[8]LOWE D A,LEPORI-BUI N,FOMIN P V,et al.Deficiency in perlecan/HSPG2 during bone development enhances osteogenesis and decreases quality of adult bone in mice[J].Calcif Tissue Int,2014,95(1):29-38.DOI:10.1007/s00223-014-9859-2.
[9]RODGERS K D,SASAKI T,ASZODI A,et al.Reduced perlecan in mice results inchondrodysplasia resembling Schwartz-Jampel syndrome[J].Human Molecular Genetics,2007,16(5):515-528.
[10]NESSLER M,PUCHALA J,KWIATKOWSKI S,et al.Multidisciplinary approach to treatment of a patient with chondrodystrophic myotonia(Schwartz-Jampel vel Aberfeld syndrome):case report and literature review[J].Ann Plast Suig,2011,67(3):315-318.DOI:10.1097/SAP.0b013e3 181fac1ec.
(本文编辑:毛亚敏)
Clinical Features of One Case of Schwartz-Jampel Syndrome Type 1A
HUANGChao1,ZHOUHai-tao1,RENXiang-yang1,MACong-min1,TENGJun-fang2*
1.DepartmentofNeurology,LuoyangCentralHospitalAffiliatedtoZhengzhouUniversity,Luoyang471000,China2.DepartmentofNeurology,theFirstAffiliatedHospitalofZhengzhouUniversity,Zhengzhou450052,China
Objective To discuss the clinical features of one child patient with Schwartz-Jampel syndrome(SJS) type 1A.Methods From June 15th to August 15th,2016,we did a review of the clinical data of one child patient with SJS type 1A from Luoyang Central Hospital Affiliated to Zhengzhou University.We summarized the patient′s main clinical features,pathological finding of biopsy specimens from the left biceps brachii muscle stained by HE stain,and results of genetic testing.Results The major clinical features of the patient were fixed facial expression,blepharophimosis,difficulty in opening eye,narrowed mouth,difficulty in opening mouth,micrognathia,slightly rigid gait and mildly elevated creatine kinase(CK) levels.Electromyogram(EMG) showed impaired myogenic response and high-frequency myotonic discharges at rest.Muscle biopsy found connective tissue slight proliferation in muscle sleeve,mildly enlarged muscle fiber diameter,a few scattered and grouped old necrotic muscle fibers with inflammatory cells infiltration,the basophilic fibers with hypertrophic nuclei,few splitted fibers and a small number of muscle fibers with nuclei immigrating to the center.The genetic analysis showed that there were compound heterozygous mutations in HSPG2 gene:c.10736T>G(p.Ile3579Ser) in exon 10,c.9963C>A(p.Cys3321Ter) in exon 73 and c.1208G>A(p.Cys403Tyr) in exon 77.Conclusion The diagnosis of SJS type 1A is determined based on the patient′s clinical features,pathological findings of muscle biopsy specimens and results of genetic testing.Special facial changes are the prominent clinical feature,and myodystrophy is the pathological change in muscles.
Osteochondrodysplasias;Myotonia; HSPG2 gene
R 681.3
B
10.3969/j.issn.1007-9572.2017.18.022
2017-01-06;
2017-03-22)
1.471000河南省洛阳市,郑州大学附属洛阳中心医院神经内科
2.450052河南省郑州市,郑州大学第一附属医院神经内科
*通信作者:滕军放,主任医师;E-mail:13838210077@163.com
*Correspondingauthor:TENGJun-fang,Chiefphysician;E-mail:13838210077@163.com
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
华东师范大学学报(自然科学版)(2017年1期)2017-02-27 13:41:06
天津农学院学报(2016年2期)2016-12-01 05:40:05
高师理科学刊(2016年8期)2016-06-15 20:27:48
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:16:04
新闻传播(2015年6期)2015-07-18 11:13:15
中国医疗美容(2015年4期)2015-04-27 02:24:06
中国医疗美容(2015年4期)2015-04-27 02:24:06
