
液相色谱串接质谱联用法确证肉食品中低浓度克仑特罗
2017-06-09 08:58:45王占良张建丽赵宇张亦农
中国运动医学杂志 2017年5期
王占良 张建丽 赵宇 张亦农
国家体育总局反兴奋剂中心食品药品兴奋剂检测实验室(北京 100029)
液相色谱串接质谱联用法确证肉食品中低浓度克仑特罗
王占良 张建丽 赵宇 张亦农
国家体育总局反兴奋剂中心食品药品兴奋剂检测实验室(北京 100029)
目的:采用液相色谱串接质谱联用法对肉食品中残留的低浓度克仑特罗进行确证分析。方法:样品匀浆处理后,酸化去除蛋白,经液液萃取和MCX 3cc固相萃取柱两步纯化,以流动相为10 mmol/L的pH 3.5甲酸铵和乙腈为流动相,梯度洗脱,采用Eclipse C18(1.8µm,4.6×100 mm)色谱柱分离,电喷雾离子源,正离子多反应监测模式扫描分析检测。结果:D9-克仑特罗为内标,克仑特罗的线性范围为0.01~0.2µg/kg,相关系数(R2)大于0.99,检出限为0.005µg/kg,3个不同水平的加标回收率为78.8%~114.8%,相对标准偏差不大于10%。结论:该方法具有操作简单、灵敏度高、重现性好等特点,可以完成肉食品样品中痕量克仑特罗的确证分析。
克仑特罗;兴奋剂;肉食品;液相色谱串接质谱联用法(HPLC-MS/MS)
克仑特罗(Clenbuterol)化学名为2-[(叔丁氨基)甲基]-4-氨基-3,5-二氯苯甲醇,一般以盐酸盐形式存在,化学结构式见图1。由于显著促进动物生长的功效,并增加瘦肉率,以及较低中毒浓度等,克仑特罗在中国畜牧业中被禁止使用,但非法使用现象时有发生。克仑特罗作为兴奋剂被世界反兴奋剂机构(World Anti-Doping Agency,WADA)列为禁止运动员使用的一种蛋白同化制剂[1],2013年1月1日,世界反兴奋剂机构(WADA)技术文件《TD2013MRPL》[2]正式实施,该技术文件规定尿样中克仑特罗的检测限由2 ng/ml变更为0.2 ng/ml,2015版技术文件依然为0.2 ng/ml的MRPL。技术文件中检测限的变化对兴奋剂检测提高了要求,同时运动员肉食品安全保障工作形势更加严峻。
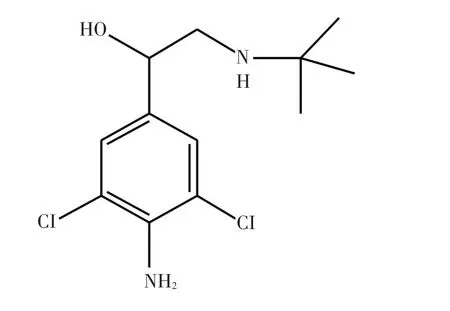
图1 克仑特罗的化学结构式
目前,肉食品和尿样中克仑特罗的检测主要有胶体金免疫层析法、气象色谱-质谱法和高效液相色谱-串接质谱法[3,4]。肉食品和尿样中现行有效国家标准方法的检测限为0.1µg/kg[5],进出口检验检疫标准方法的检测限为0.05µg/kg[6],采用液质联用高分辨质谱检测尿样中的克仑特罗可以达到pg/ml的检测水平。
经人体试验研究证实,运动员食用了克仑特罗残留浓度为0.1µg/kg的猪肉或猪内脏,就会造成运动员尿检阳性[7]。另外,人体食用食源性克仑特罗后,尿中的排泄浓度在1/2 MRPL水平可以持续7~8日[8]。因此,国家标准检测限浓度或相近浓度克仑特罗残留的肉食品对于运动员是不够安全的,无法避免运动员食源性克仑特罗问题。开发更加灵敏同时可以确证低浓度克仑特罗残留样品的检测方法很有必要。弱阳离子固相萃取柱(MCX)价格较贵,吸附能力强,在肉食品中β2-受体激动剂残留检测中应用较多,该种填料对吸附碱性物质具有良好的特异性,可以保证良好的回收率。由于填料的容量限制,在处理大容量样品时,填料的吸附能力很快饱和,回收率明显下降。
本研究采取液液萃取富集克仑特罗,并使用低填料容量MCX固相萃取柱纯化相结合的方法,有效去除基质干扰,获得良好的重现性和低浓度检测的稳定性。在检测灵敏度相对较低水平液质串接质谱联用仪上实现了克仑特罗检测的高灵敏度和良好重现性,检测限达到pg/g的水平(ng/kg)。
1 试验部分
1.1 主要仪器与装置
Agilent 1200 HPLC/6410A MS/MS液相色谱串接质谱仪:美国安捷伦公司产品,配有电喷雾离子源(ESI);Waters 20孔固相萃取装置和MCX3cc 60 mg固相萃取柱:美国沃特世公司产品;Sigma X3R高速冷冻离心机:美国西格玛奥德里奇公司产品;Ika T18分散机:广州仪科实验室技术有限公司德国伊尔姆真空泵:德国伊尔姆真空泵制造有限公司产品;Dri-block DB-3D氮气吹干装置:英国TECHNE公司产品;MILIQ纯水机:美国密理博公司产品。
1.2 主要材料与试剂
克仑特罗购买自中国食品药品检定研究院,D9-克仑特罗购买自澳大利亚标物中心,精密称取配制成1 mg/ml的甲醇溶液;氢氧化钠、氨水、高氯酸为分析纯:北京化学试剂厂产品;甲醇、甲酸铵:色谱纯,美国DIK⁃MA公司产品;50 ml螺口玻璃离心管、螺口玻璃试管、进样小瓶、进样小瓶铝盖:美国National公司产品;美国西格玛奥德里奇公司产品。
1.3 试验条件
1.3.1 色谱条件
色谱柱:Agilent Eclipse C18 100 mm×4.6 mm 1.8µm;流动相为10 mmol/L甲酸铵的水溶液和乙腈;梯度洗脱(0 min,乙腈20%;5 min,乙腈70%;7 min,乙腈75%;7.01 min,乙腈20%;);流速为0.4 ml/min;柱温15℃;进样量10µl;样品采集时间8 min。
1.3.2 质谱条件
电喷雾离子源(ESI),正离子模式,扫描方式为多反应检测方式(MRM),采集离子对见表1;干燥气流速为10 L/min;干燥气温度300℃;雾化气压力40 psi。
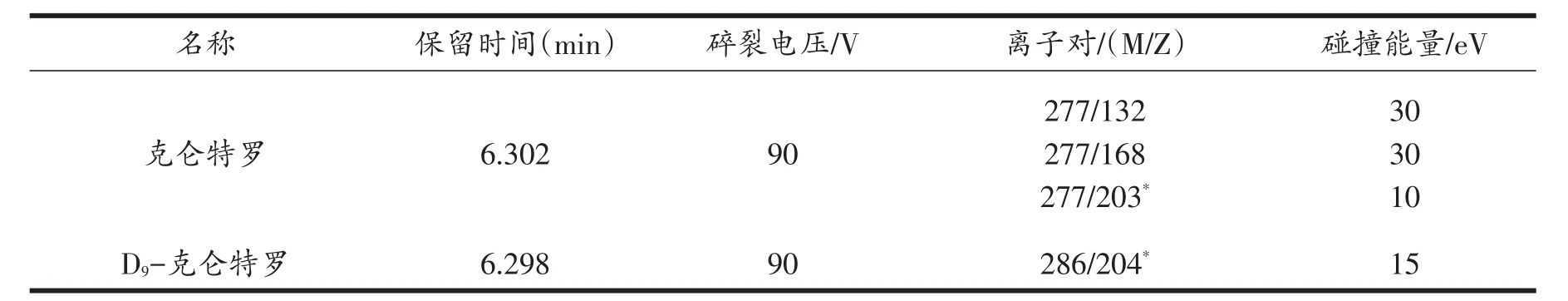
表1 克仑特罗的质谱参数
1.4 肉样前处理方法
肉样200 g,切成直径约1 cm的肉丁,准确称取10 g(误差0.2 g)置于50 ml塑料离心管内,加入质量浓度为0.1µg/L的D9-克仑特罗20µL、50%的甲醇水溶液10 ml,12000 rmp/min的转速匀浆1 min,4 ml的纯净水清洗刀头两次,合并清洗液于样品塑料离心管内,加入2 ml的浓高氯酸,超声提取30 min,温度-10℃下10000 rpm/min转速离心8 min,收集提取液于50 ml的塑料离心管内(提取液混浊时,采用滤膜滤过取出固体杂质),加入浓度12 mol/L的NaOH溶液3 ml,叔丁基甲醚10 ml,水平振荡提取10 min,温度0℃下4000 rpm/min转速离心4 min,收集上层提取液于10 ml的玻璃离心管内,65℃下氮气吹干,0.5 ml甲醇溶解残渣,加入2.5 ml的0.1 mmol/L的高氯酸,待净化。
MCX 3cc 60 mg的固相萃取柱(3 ml的5%的甲醇氨,3 ml的甲醇,3 ml的水,3 ml的0.1 mMol/L的高氯酸溶液依次活化),将玻璃离心管内溶液全部上柱,3 ml的甲醇淋洗,3 ml的5%的甲醇氨洗脱,收集洗脱液,在温度65℃下氮气吹干,加入200µL的初始比例流动相溶解残渣,待分析。
2 结果与讨论
2.1 方法优化和专属性
据文献报道[9],低浓度克仑特罗在固相萃取柱和液液萃取的两种方式的提取率有明显差异,液液萃取更加有效提取样品中低浓度克仑特罗。采用液液萃取的方式处理后,提取液的结果显示良好的回收率;但是,基质干扰很大,尤其是克仑特罗277-203的基峰离子对,有显著的增益,无法符合WADA技术文件2015ID⁃CR[10]的要求。
本实验采取药液萃取的方式对样品进行初步富集,有效提高了样品中低浓度克仑特罗的提取效率,同时结合固相萃取柱纯化,有效去除基质效应和杂质,尤其是克仑特罗基峰离子对的干扰。
称取约10 g的空白肉食品样品,添加0.005µg/kg的克仑特罗,按照1.4方法进行操作,所得到的多反应离子对监测色谱图示于图2c。结果表明,克仑特罗的特征离子对分离良好,相互间没有影响;在空白样品中,克仑特罗出峰保留时间无干扰峰出现,该方法具有较好的专属性,见图2b。
2.2 校准曲线、检测限和定量限
在空白样品中,添加分析物质量浓度为0.01~0.2 µg/kg的系列混合标准溶液,经1.4的前处理方法处理并仪器分析后,以克仑特罗的定量离子对色谱峰面积与D9-克仑特罗的定量离子色谱峰面积之比(Y)为纵坐标,分析物标准溶液的质量浓度(X)为横坐标做校准曲线,结果见表2。由表2中数据可看出,克仑特罗校准曲线相关系数平方均大于0.99,表明目标分析物在质量浓度0.01~0.2µg/kg内具有较好的线性关系,且无背景干扰。

表2 克仑特罗的线性方程、检测限和定量限
检出限(LOD)和定量限(LOQ)采用向空白样品中逐级降低加标浓度的方法来确定。以3倍信噪比(S/ N=3)对应的目标物浓度作为检出限,以S/N=10对应的目标物浓度作为定量限,获得的各目标物的检出限和定量限结果如表2所示。
2.3 回收率试验
表3是添加水平为0.01,0.02及0.1µg/kg时的回收率及精密度。由数据可以看出,分析物在高中低三种加标水平下平均回收率为78.8%~114.8%之间,相对标准偏差为6%~9%之间。本实验采用同位素内标对样品检测过程监控和定量校正,获得良好回收率,可以满足常规检测中低浓度克仑特罗样品确证的需要。
2.4 样品的确证分析
样品中克仑特罗的确证包括空白、样品3份(平行样品)、质控样品2份(空白中添加高、低浓度的克仑特罗),样品前处理程序按照1.4所述的前处理方法进行,仪器分析部分则按照1.3中方法进行,样品进样顺序采取空白、样品、空白、质控样品的进样序列进行仪器分析,质谱图结果见图2。

表3 不同添加水平下各目标物的加标回收率和相对标准偏差(RSD)(n=6)
3 结论
本研究建立了液相色谱串接质谱法确证肉食品中低浓度克仑特罗检测方法,样品采取液液萃取和固相萃取相结合的方法达到净化消除基质效应,获得良好回收率,该方法在检测灵敏度较低水平液质串接质谱联用仪上实现了克仑特罗检测的高灵敏度和良好重现性,检测限达到pg/g水平。
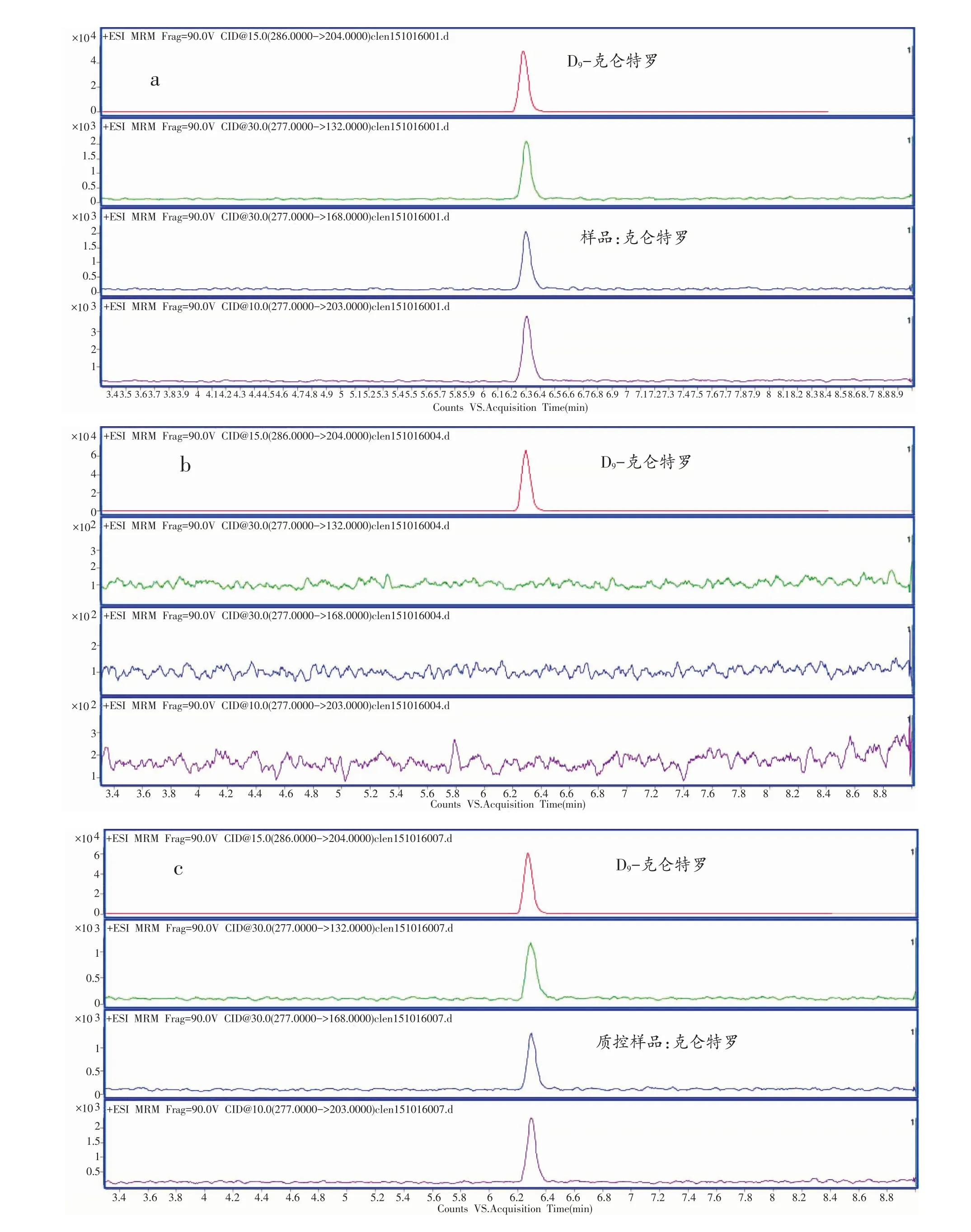
图2 克仑特罗的色谱分离图:阳性样品(a),空白样品(b)和质控样品(c)
[1] World anti-doping agency.WADA Prohibited list 2017. 2017:1-9.
[2 ]World anti-doping agency.TD2015 MRPL-Minimum Re⁃quired Performance Levels,2015:1-4.
[3]Xie Y,Chang HF,Zhao K,et al.A novel immunochromato⁃graphicassay(ICA) basedon surfaceenhancedRaman scattering for the sensitive and quantitative determination of clenbuterol.Ana.Methods,2015,7:513-520.
[4]Nicoli R,Petrou M,Badoud F,et al.Quantification of clen⁃buterol at trace level in human urine by ultrahigh pres⁃sure liquid chromatography-tandem mass spectrometry.J Chromatogr,A 1292,2013:142–150.
[5]动物源性食品中β-受体激动剂残留检测方法 液相色谱-质谱/质谱法,GBT 21313-2007.
[6]进出口动物源食品中克伦特罗、莱克多巴胺、沙丁胺醇和特布他林残留量的测定.液相色谱-质谱/质谱法,SNT 1924-2011.
[7]张建丽,王占良,高照,等.人体食源性盐酸克伦特罗尿样兴奋剂阳性可能性研究[J].中国运动医学杂志,2010,29(5):580-583.
[8]王占良,张建丽,赵宇,等.液相色谱串接质谱联用法分析人体志愿者尿样中克仑特罗[J].中国运动医学杂志,2015,34(3):309-312.
[9]Fuyu Guan,Cornelius E.Uboh,Lawrence R.Soma,et al. Quantification of clenbuterol in equine plasma,urine and tissue by liquid chromatography coupled on-line with quadrupole time-of-flight mass spectrometry.Rapid com⁃mun.Mass Spectom,2002,16:1642-1651.
[10]World anti-doping agency.WADA TD2015IDCR.2015:1-4.
Determination of Low-concentration Clenbuterol in Edible Meat Using High Performance Liquid Chromatography-tandem Mass Spectrometry
Wang Zhanliang,Zhang Jianli,Zhao Yu,Zhang Yinong
China Anti-doping Agency,Beijing 100029,China Corresponding Author:Wang Zhanliang,Email:wangzhanliang@chinada.cn
ObjectiveTo determine low-concentration clenbuterol in edible meat based on the solidphaseextraction coupled with high performance liquid chromatography-tandem massspectrometry(HPLC-MS/MS).MethodThe homogenated sample was acidized to remove proteins,and purified using the liquid-liquid extraction and MCX Oasis solid prepared extraction column,then further treated with gradientelution with themobile phaseofammonium formate(10 mmol/L and pH 3.5) and acetonitrile.The clenbuterol was completely separated on Eclipse C18(1.8µm,4.6×100 mm)column and detected in multiple reaction monitoring(MRM)mode.ResultsA good linearity was achieved for clenbuterolin the arrange of0.01~0.2 µg/kg based on the internalstandard calibration ofD9-clenbuterol,with the linearity correlation coefficient greater than 0.99 and the detection limit of 0.005 µg/kg.The relative recovery of target compounds spiked in blank sample at three levels ranging from 78.8 to 114.8%,with the relative standard deviations less than 10%.ConclusionThe method in this research is simple,rapid,reliable and suitable to confirm low-concentration clenbuterol in edible meat.
clenbuterol,doping,edible meat,high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS)
2016.01.26
王占良,Email:wangzhanliang@chinada.cn
猜你喜欢
资源节约与环保(2022年8期)2022-09-20 02:25:58
成都体育学院学报(2021年1期)2021-07-16 07:37:32
基层中医药(2020年8期)2020-11-16 00:55:14
畜牧兽医科学(2018年3期)2018-02-15 20:08:15
发明与创新(2016年33期)2016-08-21 13:22:20
湖南畜牧兽医(2016年1期)2016-06-05 08:37:49
兽医导刊(2016年6期)2016-05-17 03:50:59
甘肃畜牧兽医(2016年17期)2016-03-12 04:15:34
中国药业(2014年19期)2014-05-17 03:12:26
中国氯碱(2014年11期)2014-02-28 01:05:04
