
江汉油田区典型农田土壤烃类降解微生物功能基因bssA的遗传多样性研究*
2017-06-07 10:30:36左小虎王明霞姚炎红李振轮周志峰
土壤学报 2017年3期
左小虎 王明霞 姚炎红 李振轮 周志峰
(西南大学资源环境学院,重庆 400716)
江汉油田区典型农田土壤烃类降解微生物功能基因bssA的遗传多样性研究*
左小虎 王明霞 姚炎红 李振轮 周志峰†
(西南大学资源环境学院,重庆 400716)
油田区土壤易受烃类物质影响并可能富集了特异的石油烃降解微生物类群。针对江汉油田区5个不同油井口附近的典型旱地农田土壤,采用石油烃(Petroleum hydrocarbons,PHs)中苯系物代谢的关键功能基因-bssA(苯甲基琥珀酸合成酶基因)作为分子标识物,通过克隆文库结合末端限制性片段长度多样性(Terminal-restriction fragment length polymorphism,T-RFLP)的方法,研究该油田区土壤含有bssA基因的烃类降解微生物群落结构,并探讨其环境驱动机制。结果表明,土壤中PAHs含量在0.21~2.01 mg kg-1之间,石油烃污染程度较低。T-RFLP的分析表明不同土壤样品中的bssA基因多样性差异明显,PAHs(Polycyclic Aromatic Hydrocarbons,多环芳烃)含量最高土壤中bssA基因多样性最高,其优势bssA基因类群与硫酸盐还原菌或地杆菌有较近的亲缘关系。冗余分析进一步表明,土壤硝态氮、有效磷、PAHs含量均是影响bssA基因多样性的重要因子。这些结果表明:江汉油田区典型农田土壤中含有bssA基因的主要类群为β-变形菌和δ-变形菌,并与地杆菌属(Geobacter)、索氏菌属(Thauera)和固氮菌属(Azoarcus)具有较近的系统发育亲缘关系。这些微生物可能通过硝酸盐、硫酸盐及铁还原代谢过程降解土壤PAHs。
土壤;厌氧降解;苯甲基琥珀酸合成酶基因(bssA);群落结构
石油烃(Petroleum hydrocarbons,PHs)污染是油田开发面临的重要问题,尤其是油田区域的农田土壤,其污染物微生物修复得到了广泛关注。石油烃是由烷烃、烯烃、芳香烃、杂环芳烃等多组分的复杂分子组成的均质混合物,其中的多环芳烃(Polycyclic Aromatic Hydrocarbons,PAHs)和苯系物(苯、甲苯、乙苯、二甲苯等)备受关注,是土壤中常见的有机污染物[1-3],不仅严重危害土壤环境质量,也会直接或间接危害人类健康[4-5]。因此,土壤中PAHs和苯系物降解微生物一直是学术界关注的热点[6-7]。一般认为,在纯培养体系有氧条件下PAHs及苯系物的微生物降解更易发生,但在复杂土壤和沉积物等自然环境中,石油烃大多闭蓄于微孔介质等厌氧或微氧生境,厌氧微生物降解可能发挥了重要的作用[3,8]。例如,在苯系物、烃类及萘等低分子量PAHs的厌氧降解过程中,由甘氨酰自由基合成酶所催化的延胡索酸加入途径起着十分重要的纽带作用[9]。该途径最早发现于β-变形菌厌氧降解甲苯的过程中,该过程中的关键环节是由苯甲基琥珀酸合成酶(Benzylsuccinate synthase,BSS)基因所编码的BBS催化延胡索酸加入到甲苯[10],形成的(R)-苯甲基琥珀酸酯[11-13]可经随后的一系列反应最终转化为关键中间代谢物苯甲酰-CoA[9]。随后的研究表明,该反应普遍存在于苯系物,烷烃及PAHs的厌氧降解过程中。如,硫酸盐还原菌(Sulfate-reducing bacteria,SRB)和硝酸盐还原菌(Nitrate-reducing bacteria,NRB)在厌氧条件下也可以对n-烷烃进行类似于甲苯添加延胡索酸的活化降解,该过程由烷基琥珀酸合成酶(ASS)催化完成[14-15],而SRB则是通过萘基-2-甲基-琥珀酸合成酶(NMS)催化延胡索酸加入2-甲基萘[16-17],来完成厌氧代谢2-甲基萘的第一步反应。因此,近年来,编码BSS,ASS及NMS的关键功能基因(bssA;assA;nmsA)被尝试作为分子标识物,研究环境中苯系物等PHs类化合物厌氧代谢的微生物类群[10,18-19],表征环境受污染程度及微生物降解的潜在能力。如,Frederick等[20]以bssA、nmsA和assA作为特异性标识物,研究了一些石油烃污染环境(陆地及海洋)中的厌氧PHs降解菌群,并发现了一些与16S rRNA基因分类迥然不同的δ-变形菌和梭菌类群。Alejandro 等[21]比较了bssA、nmsA和assA基因的遗传多样性,研究了原油泄漏区域海水及沉积物中PHs厌氧降解菌的群落结构,发现这些基因的丰度及多样性具有明显的差异,并与受污染的程度密切相关。但已有的研究均表明,与nmsA和assA分子标靶基因相比较,以bssA为代表的编码延胡索酸加入的关键基因具备较强的专一性,是未来研究石油污染环境微生物修复的重要标识物。但目前相关的bssA基因研究多见于海洋石油污染环境,油田区土壤中可能富集了大量的石油烃类降解微生物[7,22],研究其中的关键功能基因多样性对污染环境生物修复具有重要意义。
油田区域的土壤有受原油污染的风险,而土壤中极可能存在着厌氧/微氧环境[23]。据此,本研究针对具有50年油气开发历史的江汉油田区,在约20 km2的范围内,选择了5个不同油井附近的典型旱地农田土壤,以bssA基因为分子标识物,通过克隆文库结合T-RFLP的方法研究了油田区典型农田土壤中苯系物厌氧降解微生物的群落结构,初步探讨了其群落结构与环境因子之间的关系,为进一步深入研究PHs类有机污染物的土壤厌氧代谢的微生物机理提供了一定的理论依据。
1 材料与方法
1.1 土壤样品采集
江汉油田位于湖北省潜江市境内,该油田已有50多年的油气开发历史,是新中国最早开发的油气田之一。本研究的土壤样品采集于2014年7月。在江汉油田区域共选择5个不同的油井,编号为JH-1 至JH-5,每个油井口30~40 m半径范围内,采集农田0~20 cm表层新鲜土壤(潮土,黏壤)。在每个采样点中心周围30 m2的范围内,随机选择9个点,进行多点混合采样并初步剔除石块和杂物,以完成单个土样的采集,且各样点内均按上述方法采集3个重复的土壤样品。采集好的样品装于自封袋后,置于放有冰袋的箱子中运回实验室。随后,过2 mm筛后分装,部分放于4 ℃用于基本性质测定,部分置于-20 ℃冰箱用于DNA的提取。
1.2 土壤性质及PAHs含量测定
土壤pH采用2.5∶1的水土比,用“PHS-3C型”pH仪测定。土壤总有机质、有效磷、铵态氮和硝态氮的测定参照《土壤农业化学分析方法》[24]进行,分别使用重铬酸钾氧化还原滴定法(K2Cr2O7-H2SO4法)、钼锑抗比色法、靛酚蓝分光光度法和萘乙二胺分光光度法测定。
土壤中PAHs含量的测定采用经典的索氏提取法[25]。具体步骤如下:称取10 g经冷冻干燥的土壤样品、10 g无水硫酸钠及少量铜粉置于同一烧杯中混匀,无损移入滤纸桶,加入150 ml丙酮–正己烷(1∶1)混合液浸泡土壤样品12 h,将滤纸桶装入索氏提取器中75 ℃提取6 h;提取液于-46 kPa,45 ℃下旋转蒸发浓缩至1~2 ml,然后用硅胶层析柱(1 cm无水硫酸钠,6 cm活化硅胶和3 cm氧化铝)净化;以15 ml色谱级正己烷预洗后,用70 ml二氯甲烷和正己烷的混合液(3∶7,V/V)洗脱;收集洗脱液,洗脱液旋转蒸发至1~2 ml,再加入25 ml甲醇进行溶剂置换,再旋转蒸发浓缩并用甲醇定容至1 ml;最后用高效液相色谱仪(HITACHI L-7100,日本)测定各样品中PAHs含量。
1.3 土壤DNA提取
土壤DNA提取:使用PowerSoil®DNA Isolation Kit(MO BIO,美国)试剂盒提取土壤DNA。称取0.5 g土壤样品,具体操作步骤按说明书进行。提取到的总DNA,用1%的琼脂糖凝胶电泳检测提取的DNA产物,并用DU 800 spectrophotometer (Beckman Coulter Inc,美国)测定其浓度,最后置于-20 ℃保存备用。
1.4 克隆文库构建及T-RFLP分析
为了全面解析所采土壤样品中bssA、nmsA和assA基因的类群,将所有5个土壤样品的bssA、nmsA和assA基因PCR扩增产物分别混合并构建其混合克隆文库,为后续T-RFLP分析中限制性内切酶的选择提供依据,同时也有助于确定T-RFLP分析中所获取的各末端片段所对应的类群。具体步骤如下:分别以5个不同土壤样品的DNA为模板,利用表1中的引物及相应的扩增条件对上述各目标基因进行扩增,聚合酶链式反应主要包括:0.25 μl TaKaRa Ex Taq(5 U μl-1)、2.5 μl 10×Ex Taq Buffer、2 μl dNTP Mixture(2.5 m mol-1)、前后引物各0.5 μl(10 μm mol-1)、2 μl DNA模板,加入灭菌水补足至25 μl反应体系。将不同土壤样品bssA、nmsA及assA基因的PCR产物分别混合,混合后的PCR产物用Wizard SV Gel and PCR Clean-Up System(Promega,美国)进行纯化,纯化后的PCR产物与pGEM-T Easy Vector(Promega,美国)连接,之后转入Escherichia coli JM109 (TakaRa,日本)感受态细胞,通过蓝白斑筛选,各自随机挑选阳性克隆子送上海生物工程有限公司进行测序,从而完成各目的基因混合克隆文库的构建。

表1 PCR扩增引物及反应程序Table 1 Primer sets and PCR procedures
使用DNAMAN(Version 6.0.3.48,Lynnon Biosoft,美国)对所得序列进行同源性分析,相似度>9 5%的序列归入同一分类操作单元(Operational Taxonomic Unit,OTU),每个OTU选择一条具有代表性的序列,采用邻位相邻法(Neighbor-Joining),用MEGA(version 6.0)软件构建系统发育树。之后,使用DNAMAN软件对各OTU的代表性序列进行酶切位点分析,结果表明限制性内切酶Alu I(Takara,日本)能够较好地对bssA基因克隆文库中优势的OTU进行分型,可作为后续T-RFLP分析中的限制性内切酶。本研究所获得的序列已提交至GenBank并获取了相应的序列号(KX148522-KX148545)。据此,进一步分别以各土壤样品的DNA为模板,用前引物5′端带FAM标记的引物对(7772f/8546r)对bssA基因进行PCR扩增。获得的PCR产物纯化后,用Alu I在37 ℃酶切4 h,酶切产物用醋酸钠和冰乙醇纯化[26],随后用核酸测序仪ABI PRISM3700(Applied Biosystems,美国)进行基因扫描,用GeneMapper (Applied Biosystems,美国)软件对T-RFLP图谱进行分析。
1.5 数据处理
所有数据采用SPSS 17.0进行处理分析,样品之间的平均值差异采用One–Way ANOVA单因素方差分析,p<0.05表示差异显著。用CANOCO (4.5)中的冗余分析(Redundancy analysis,RDA)对bssA基因多样性与环境因子之间的关系进行分析。
2 结 果
2.1 土壤PAHs含量及理化性质
不同油田附近典型农田土壤PAHs含量及基本性质见表2。各土壤样品PAHs含量具有明显差别,其中JH-5(2.01 mg kg-1)和JH-1(1.23 mg kg-1)的含量较高,其余各样品均低于1 mg kg-1。供试土壤均呈碱性,且各样品pH之间的差异性不显著。不同土壤样品的有机质含量差异性显著(p <0.05),其中样品JH-5和JH-2的有机质含量较高,分别为40.2 g kg-1和38.3 g kg-1,而JH-9的有机质含量最低,仅为11.3 g kg-1。有效磷含量最高的样品是JH-5(25.4 mg kg-1),其次是JH-3(15.9 mg kg-1)和JH-4(11.5 mg kg-1),其余各样品的有效磷含量均低于10 mg kg-1。各土壤样品的铵态氮含量差异不明显,均在4.10~4.30 mg kg-1之间。硝态氮含量最高的是JH-5(31.1 mg kg-1),其次是JH-3和JH-4,其余样品均低于5 mg kg-1。
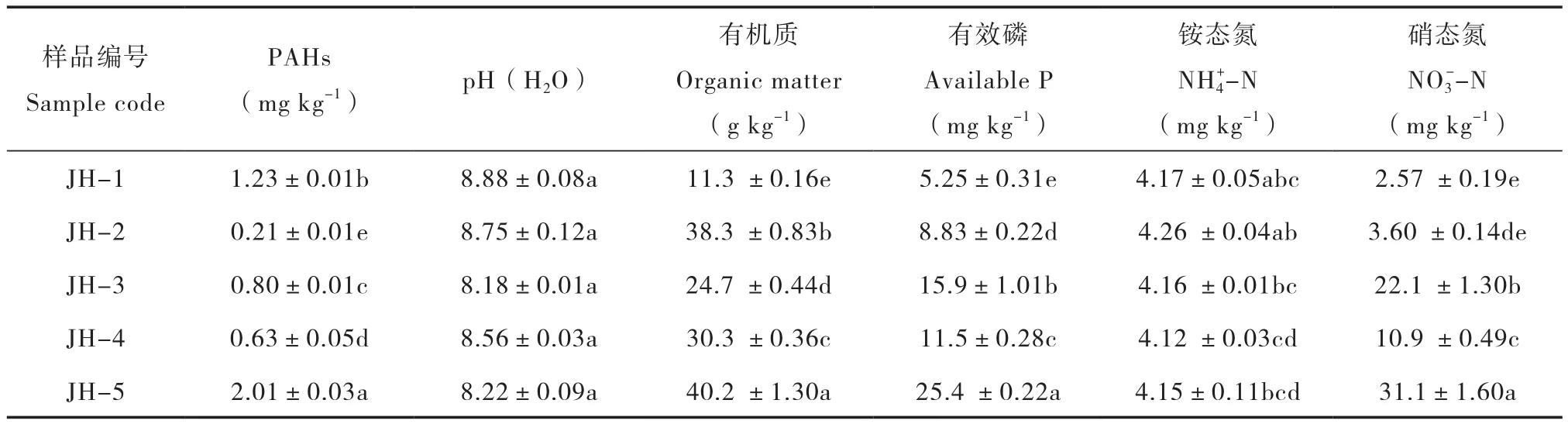
表2 样点土壤基本性质Table 2 Basic properties of the tested soil samples
2.2 混合克隆文库中bssA序列及其末端限制性片段组成
对bssA、nmsA和assA克隆测序结果进行比对分析,结果表明只成功扩增到土壤样品中的bssA基因。从bssA混合克隆文库中随机挑选220个阳性克隆子测序,并在GenBank中进行Blast比对,共得到95条有效的bssA序列。在95%的相似水平下,可分为24个OTUs。各OTU代表性序列经Alu I酶切后的末端限制性片段(Terminal-restriction fragment,T-RF)长度及其所含序列占克隆文库总序列数的百分比如下表3所示。可见,其序列数占克隆文库总序列数的比例超过10%的优势OTUs为OTU-3 (10.5%)、5(11.55%)、6(17.85%)和23 (13.65%)。从表中还可看出,限制性内切酶Alu I能够较好地将这些优势OTUs进行分型,适于后续的T-RFLP分析。
从每个OTU中选择一条代表性的序列来构建bssA基因的系统发育树(图1)。从图1可知,该油田区典型农田土壤bssA基因可分为4个不同的簇(Clusters1-4)。其中,Cluster1含有8个OTUs,且包括占优势的OTU-3(571bp T-RF)和OTU-6 (409bp T-RF),它们与β-变形菌有较近的亲缘关系。而Cluster2和Cluster4中各含有2个和4个OTUs,其所含序列数分别占克隆文库总序列数的4.2%和11.2%。其余的10个OTUs 均属于Cluster3,包含占据优势的OTU-5(388bp T-RF)和OTU-23(433bp T-RF)。此外,Cluster2、 Cluster3 和 Cluster4均与δ-变形菌的亲缘关系较近。通过对bssA基因混合克隆文库的构建及相应的系统发育分析,可为后续T-RFLP中确认不同长度末端限制性片段(T-RFs)所代表的具体bssA类群提供依据。
2.3 土壤bssA基因的遗传多样性及其影响因子
各样品bssA基因的T-RFs组成如图2所示。由图可知,样品JH-1和JH-2中bssA的群落组成较为相似,均含有3种T-RFs类群(96、154、409 和433bp),且409bp和433bp的T-RFs是其共有的优势类群。结合2.2的结果可知,409bp T-RF可能属于OTU-1、OTU-6或OTU-18,在系统发育树中位于Cluster1,与β-变形菌门的红环菌科(Rhodocyclaceae)下的索氏菌属(Thauera)和固氮弓菌属(Azoarcus)有较近的亲缘关系。而433bp的T-RFs则属于OTU-23,位于Cluster3,与δ-变形菌门的地杆菌科(Geobacteraceae)的地杆菌属(Geobacter)有较近的亲缘关系。样品JH-3中只检测到一个252bp(不确定类群)独特的bssA类群。样品JH-4中的bssA由89bp(Cluster2或4)、175bp(Cluster3)、205bp(不确定类群)和295bp(不确定类群),共4种T-RFs所组成。
而在PAHs含量最高的JH-5中,含有的T-RFs类群最多,即89bp(Cluster4)、96bp(不确定类群)、175bp(Cluster3)、205bp(不确定类群)、295bp(不确定类群)、388bp(Cluster2或3)、409bp(Cluster1)及433bp(Cluster3),共8种T-RFs。其中388bp T-RF(Cluster2或3,硫酸盐还原菌或地杆菌)的类群是其特异性类群。值得注意的是,在PAHs含量较高的JH-1和JH-5中,尽管bssA基因的组成差异明显,但是组成JH-1中3种bssA类群(96,409和433bp)均出现在后者中,且96bp(不确定类群)是其所独有的T-RFs。由此可见,96bp及388bp 的bssA类群可能受到土壤PAHs的富集。将土壤基本性质及T-RFLP图谱(去除过于单一的JH-3)分别作为环境变量和物种变量,对土壤环境因子与bssA群落结构进行冗余分析的结果(图3)表明,四个样品的bssA基因群落组成差异明显,均不在一个象限。此外,除有效磷、硝态氮、铵态氮等因子外,土壤PAHs含量也是影响bssA群落组成的关键因子之一,且96bp(不确定类群)和388bp(硫酸盐还原菌或地杆菌)T-RFs所代表的bssA类群与PAHs含量呈正相关。
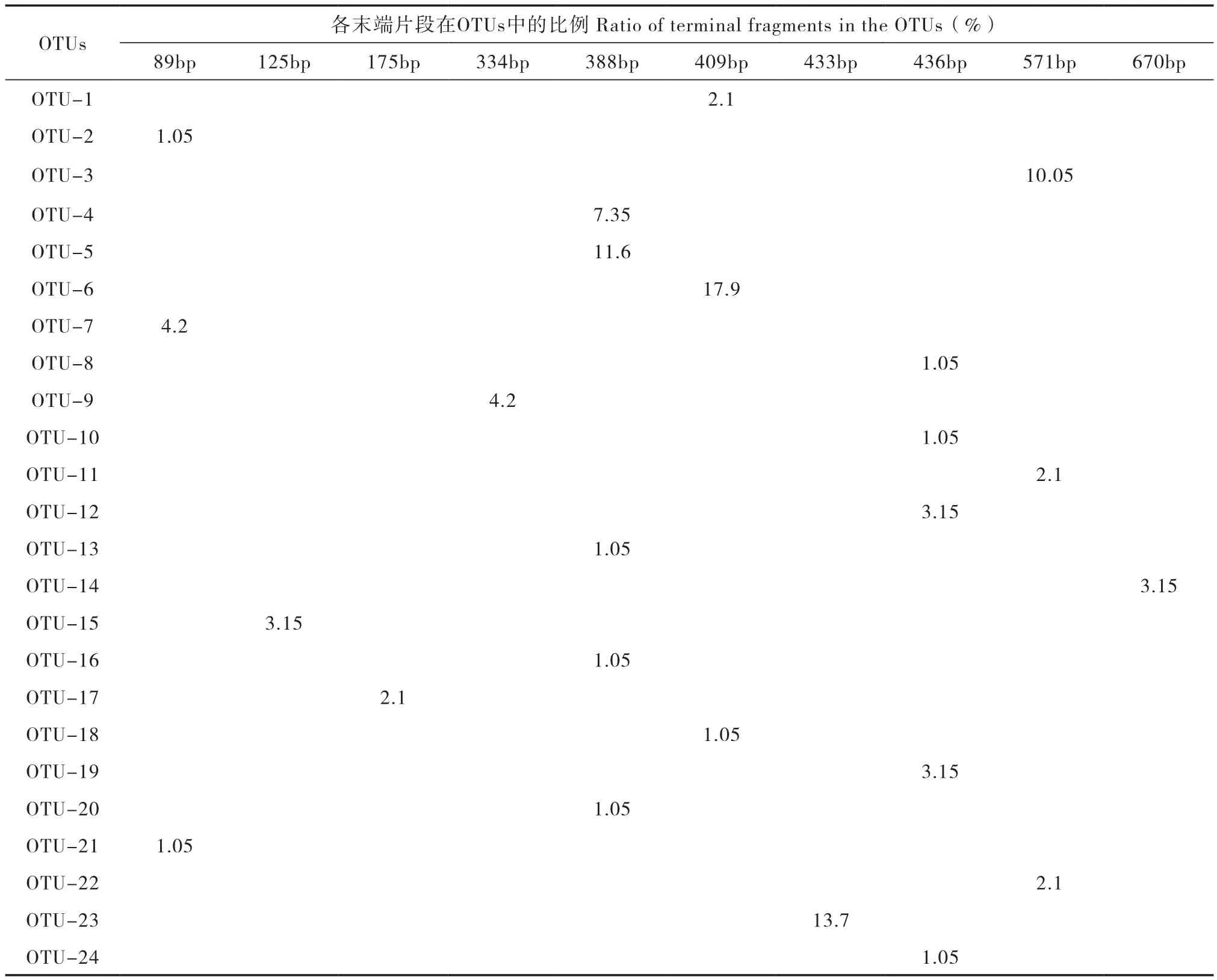
表3 克隆文库中的OTUs及Alu I 酶切的末端片段Table 3 OTUs in the bssA clone library and Alu I-digested TRFs
3 讨 论
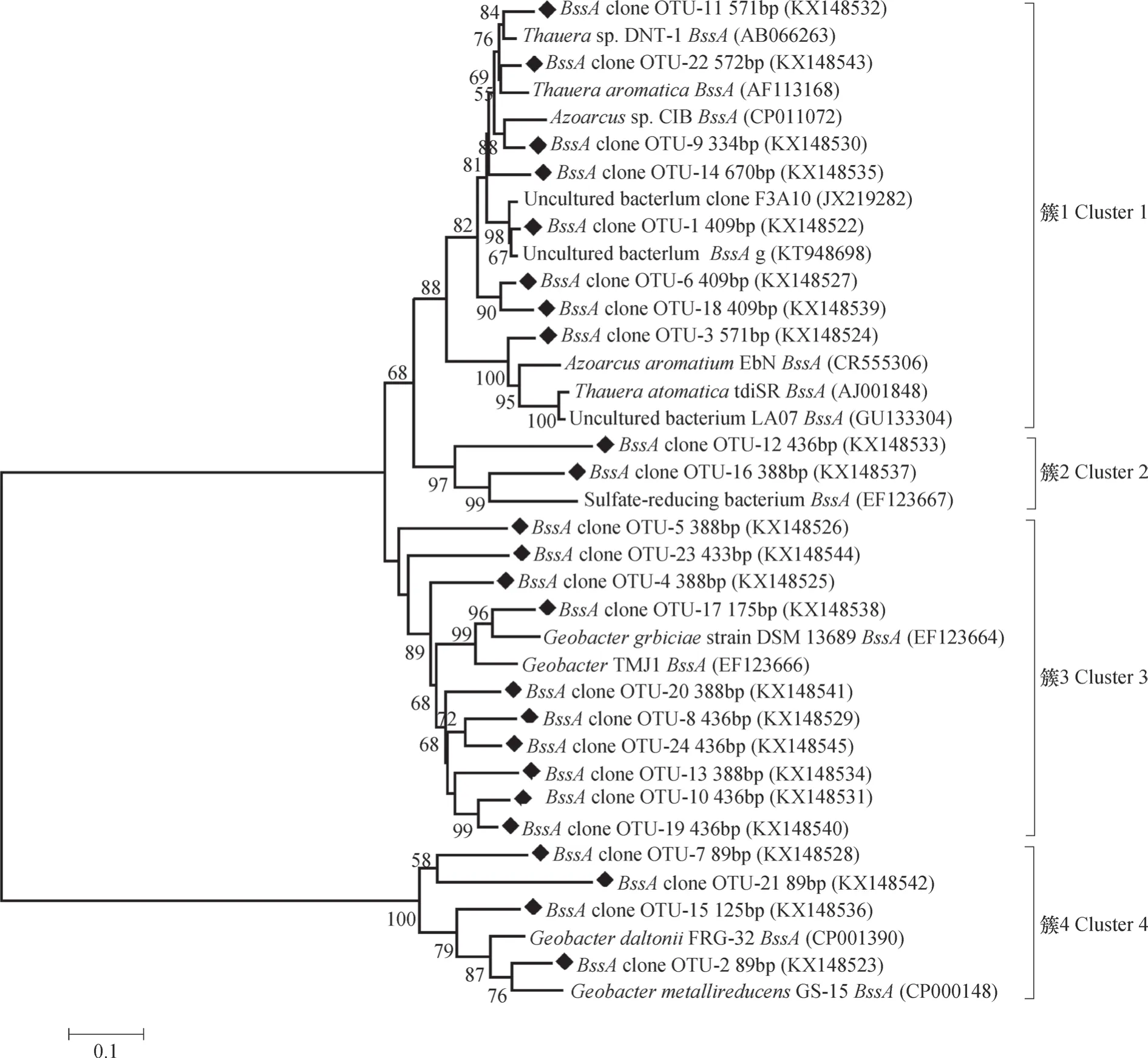
图1 基于bssA克隆文库构建的N-J(Neighbor-joining)系统发育树Fig. 1 Phylogenetic tree based on bssA clone library
本研究中,土壤样品的PAHs含量范围为0.21~2.01 mg kg-1,低于中华人民共和国环境保护部2008年颁布的《土壤环境质量标准》修订版(征求意见稿)中拟定的农业用地土壤16种优先控制PAHs总含量的筛选值(4.90~10.1 mg kg-1)[27]。据报道,长江上游重庆金佛山土壤16种优先控制PAHs的含量范围为0.24~2.12 mg kg-1[28]。而对位于长江下游长江三角洲的苏州市土壤PAHs含量的调查表明,该区域土壤总PAHs含量范围为0.045~3.70 mg kg-1[29]。由此可见,本研究各样点土壤中PAHs含量均在上述调查结果的范围之内,虽源于油井口附近,却未遭受严重的污染。同时检测到了苯系物厌氧代谢的关键基因-bssA,表明这些农田土壤具有降解石油烃污染物的遗传基础。克隆文库的结果表明,该油田区土壤bssA类群主要为β-变形菌(Cluster1)和δ-变形菌(Cluster2、Cluster3和Cluster4),它们所含序列数分别占克隆文库总序列数的42.6%和57.4%。已有的研究表明,变形菌门是多数PHs污染土壤中的优势bssA类群[20-21],这与本研究的结论一致。其中,β-变形菌门的一些反硝化细菌可以芳香烃为唯一碳源进行生长[30-31]。而δ-变形菌中的一些硫酸盐还原菌、铁还原菌等也具备厌氧代谢芳香烃的能力[32]。从属的水平来看,江汉油田区土壤中β-变形菌的bssA类群与索氏菌属(Thauera)和固氮弓菌属(Azoarcus)有较近的亲缘关系,它们均具备在反硝化条件下厌氧代谢芳香烃化合物的能力[33]。而该区域土壤中δ-变形菌的bssA类群则与硫酸盐还原菌TRM1(Sulfate-reducing bacterium TRM1)及地杆菌属(Geobacter)有较近的亲缘关系,上述两种菌常见于PHs污染土壤中,它们分别能够以硫酸根及铁离子为电子受体在厌氧条件下降解芳香烃化合物[34]。如,Winderl等[18]报道,德国慕尼黑煤气厂附近的水体沉积物中的主要bssA类群为地杆菌属。Sun等[22]则在分别添加了硝酸盐和硫酸盐的甲苯污染土壤中,分别检测到索氏菌DNT-1(Thauera sp. DNT-1)和硫酸盐还原菌PRTOL1(sulfate-reducing bacterium PRTOL1)。此外,据报道,δ-变形菌N47和 NaphS2均含有nmsA基因[17,20],而脱硫肠杆菌属(Desulfotomaculum)的一些菌株则含有assA基因[19]。本研究中含有bssA基因的类群,目前还没有发现它们同时含有nmsA或assA基因,这可能是后两种基因不能成功从该区域土壤中成功扩增的原因。
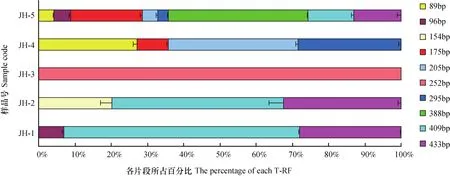
图2 土壤样品中bssA群落结构的组成Fig. 2 Structural composition of the bssA community in the soil samples
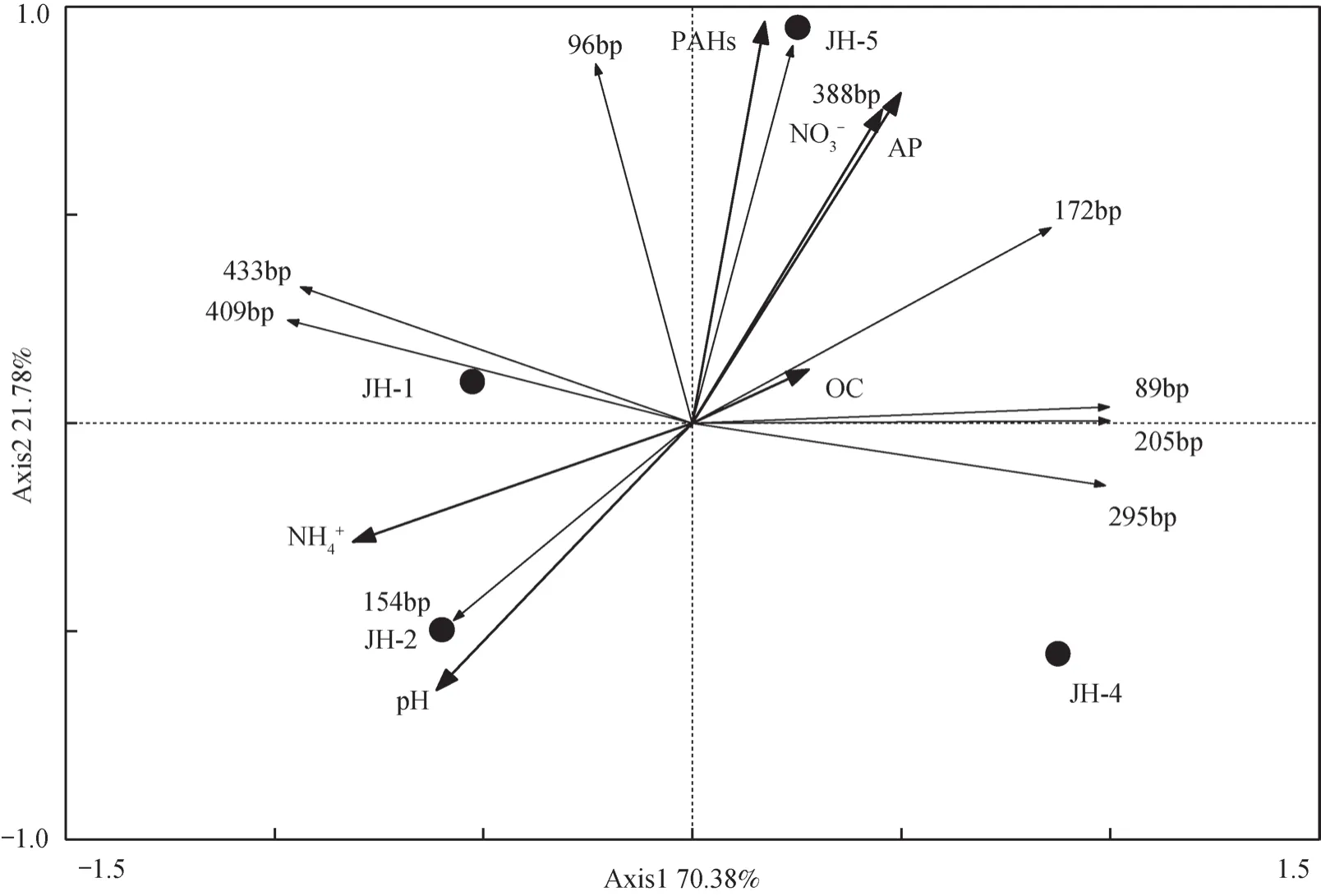
图3 基于bssA基因T-RFLP剖面与土壤性质的RDA分析Fig. 3 RDA analysis based on T-RFLP of bssA and soil properties
本研究中T-RFLP的结果(图2)表明,该油田区域不同样点土壤样品中的bssA微生物群落结构差异明显。其中,在PAHs含量最高JH-5中,具有最多的bssA基因类群,且388bp(Cluster2或3,硫酸盐还原菌或地杆菌)是其特异性的优势T-RFs,这可能表明较高含量的PHs有机污染物对土壤中硫酸盐还原菌或铁还原菌具有一定富集作用。Alejandro等[21]也发现,海洋沉积物中受原油污染程度越大、时间越久,bssA基因的多样性越高。结合上述分析可知,硫酸盐还原或铁还原条件可能是重度PHs污染土壤中污染物去除的重要厌氧代谢途径[31]。在PAHs含量仅次于JH-5的JH-1中,虽然仅检测到3种bssA类群,但它们均出现在JH-5的bssA类群中,且96bp(不确定类群)的bssA类群只出现在它们中,这可能体现出PHs类污染物对土壤bssA类群的选择性。此外,在PAHs含量较低的样品中也出现了上述优势bssA类群,如409bp和433bp 也是JH-2的优势bssA类群,而JH-4的bssA类群多于JH-1,且其bssA类群均出现在JH-5中。可能原因有以下两点:首先,所有的土壤样品均采自同一区域,且类型相同,因此其土著微生物群落结构(包含bssA类群)也应该具有一定的同源性。其次,除PAHs外,土壤的其他理化性质,如NO3-、NH4+、有效磷等也会对微生物的群落结构产生影响[35]。JH-3中仅检测到一个bssA类群,这可能是由于某些未知的土壤环境因子或人为干扰所导致。随后的RDA分析(图3)进一步表明,除能够作为微生物生长所必须的铵态氮、硝态氮及有效磷外,土壤PAHs含量也是影响bssA群落结构的重要环境因子。与T-RFLP结果一致的是,96bp(不确定类群)和388bp(硫酸盐还原菌或地杆菌)T-RFs所代表的bssA类群与土壤PAHs含量呈正相关,表明PHs污染可能对它们有较强的富集作用。综上所述,江汉油田区域土壤含有PHs类污染物厌氧降解的遗传基础,可能通过不同的细菌在多种代谢途径下进行,如以硝酸盐为电子受体的反硝化作用,以硫酸盐为电子受体的硫酸盐还原作用及以铁离子为电子受体的铁还原作用等。同时,土壤环境条件及受污染程度均可能影响bssA类群及其对PHs的降解效率。
4 结 论
本研究以典型PHs类污染物-苯系物厌氧代谢的功能基因bssA作为标识,通过克隆文库结合T-RFLP的方法,研究了典型油田区域土壤
PHs降解微生物的遗传多样性。该油田区主要的bssA类群为β-变形菌和δ-变形菌,包括索氏菌属(Thauera)、固氮弓菌属(Azoarcus)、硫酸盐还原菌以及地杆菌属(Geobacter),已知的这些微生物具有硝酸盐还原、硫酸盐还原及铁还原代谢能力,可能是江汉油田区土壤厌氧微域中PHs降解的主要微生物类群。土壤氮、磷及PHAs含量均是影响bssA基因遗传多样性的关键因子,较高含量的PAHs可能对含有bssA基因的硫酸盐还原菌及地杆菌属的微生物具有一定的富集作用。
[1] Anderson R T,Lovley D R. Ecology and biogeochemistry of in situ groundwater bioremediation. Advances in Microbial Ecology,1997,15:289—350
[2] Lin B,van Verseveld H W,Röling W F. Microbial aspects of anaerobic BTEX degradation. Biomedical and Environmental Sciences,2002,15(2):130—144
[3] Chakraborty R,Coates J D. Anaerobic degradation of monoaromatic hydrocarbons. Applied Microbiology and Biotechnology,2004,64(4):437—446
[4] Cai Q Y,Mo C H,Li Y H,et al. Occurrence and assessment of polycyclic aromatic hydrocarbons in soils from vegetable fields of the Pearl River Delta,South China. Chemosphere,2007,68(1):159—168
[5] Puglisi E,Cappa F,Fragoulis G,et al. Bioavailability and degradation of phenanthrene in compost amended soil. Chemosphere,2007,67(3):548—556
[6] Heider J,Spormann A M,Beller H R,et al. Anaerobic bacterial metabolism of hydrocarbons. FEMS Microbiology Reviews,1998,22(5):459—473
[7] Kleinsteuber S,Schleinitz K M,Vogt C. Key players and team play:Anaerobic microbial communities in hydrocarbon-contaminated aquifers. Applied Microbiology and Biotechnology,2012,94(4):851—873
[8] Grossi V,Cravo L C,Guyoneaud R,et al. Metabolism of n-alkanes and n-alkenes by anaerobic bacteria:A summary. Organic Geochemistry,2008,39(8):1197—1203
[9] Heider J. Adding handles to unhandy substrates:Anaerobic hydrocarbon activation mechanisms. Current Opinion in Chemical Biology,2007,11(2):188—194
[10] Beller H R,Kane S R,Legler T C,et al. A realtime polymerase chain reaction method for monitoring anaerobic,hydrocarbon-degrading bacteria based on a catabolic gene. Environmental Sciences andTechnology,2002,36(18):3977—3984
[11] Biegert T,Fuchs G,Heider J. Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. European Journal of Biochemisty,1996,238(3):661—668
[12] Beller H R,Spormann A M. Anaerobic activation of toluene and o-xylene by addition to fumarate in denitrifying strain T. Journal of Bacteriology,1997,179(3):670—676
[13] Leuthner B,Leutwein C,Schulz H,et al. Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica:A new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Molecular Microbiology,1998,28(3):615—628
[14] Rabus R,Wilkes H,Behrends A,et al. Anaerobic initial reaction of n-alkanes in a denitrifying bacterium:Evidence for(1-methylpentyl)succinate as initial product and for involvement of an organic radical in n-hexane metabolism. Journal of Bacteriology,2001,183(5):1707—1715
[15] Kropp K G,Davidova I A,Suflita J M. Anaerobic oxidation of n-dodecane by an addition reaction in a sulfate-reducing bacterial enrichment culture. Applied and Environmental Microbiology,2000,66(12):5393—5398
[16] Selesi D,Jehmlich N,Bergen M,et al. Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. Journal of Bacteriology,2010,192(1):295—306
[17] Musat F,Galushko A,Jacob J,et al. Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environmental Microbiology,2009,11(1):209—219
[18] Winderl C,Schaefer S,Lueders T. Detection of anaerobic toluene and hydrocarbon degraders in contaminated aquifers using benzylsuccinate synthase (bssA)genes as a functional marker. Environmental Microbiology,2007,9(4):1035—1046
[19] Callaghan A V,Davidova I A,Savage-Ashlock A K,et al. Diversity of benyzl- and alkylsuccinate synthase genes in hydrocarbon-impacted environments and enrichment cultures. Environmental Science and Technology,2010,44(19):7287—7294
[20] Frederick V N,Giovanni P,Sara K,et al. Enhanced gene detection assays for fumarate-adding enzymes allow uncovering of anaerobic hydrocarbon degraders in terrestrial and marine systems. Applied and Environmental Microbiology,2013,79(2):543—552
[21] Alejandro A G,Ramon R M,Silvia M. Diversity of benzylsuccinate synthase-like(bssA)genes in hydrocarbon-polluted marine sediments suggests substrate-dependent clustering. Applied and Environmental Microbiology,2013,79(12):3667—3676
[22] Sun W M,Sun X X,Alison M,et al. Presence,diversity and enumeration of functional genes(bssA and bamA)relating to toluene degradation across a range of redox conditions and inoculum sources. Biodegradation,2014,25(2):189—203
[23] Johnsen A R,Wick L Y,Harms H. Principles of microbial PAH-degradation in soil. Environmental Pollution,2005,133(1):71—84
[24] 鲁如坤. 土壤农业化学分析方法. 北京:中国农业科技出版社,2000
Lu R K. Analytical methods for soil and agro-chemistry (In Chinese). Beijing:China Agricultural Science and Technology Press,2000
[25] 赵有政,裔兆军. 江苏盐城滩涂土壤中多环芳烃的测定. 污染防治技术,2011,24(5):41—43
Zhao Y Z,Yi Z J. Determination of polycyclic aromatic hydrocarbons(PAHs)in the Coastal soil of Yancheng in Jinagsu Province(In Chinese). Pollution Control Technology,2011,24(5):41—43
[26] Chen X,Zhang L M,Shen J P,et al. Abundance and community structure of ammonia-oxidizing archaea and bacteria in an acid paddy soil. Biology and Fertility of Soils,2011,47(3):323—331
[27] 中华人民共和国环境保护部. 土壤环境质量标准(征求意见稿). GB15618-2008,2008
Ministry of Environmental Protection of the People's Republic of China. Environmental quality standard for soils(exposure draft)(In Chinese). GB15618-2008,2008
[28] 师阳,孙玉川,梁作兵,等. 重庆金佛山土壤中PAHs含量的海拔梯度分布及来源解析. 环境科学,2015,36(4):1417—1424
Shi Y,Sun Y C,Liang Z B,et al. Altitudinal gradient distribution and source analysis of PAHs content of topsoil in Jinfo Mountian,Chongqing(In Chinese). Environmental Science,2015,36(4):1417—1424
[29] 刘增俊,腾应,黄标,等. 长江三角洲典型地区农田土壤多环芳烃分布特征与源解析. 土壤学报,2010,47 (6):1110—1117
Liu Z J,Teng Y,Huang B,et al. Distributionand sources analysis of PAHs in farmland soils in areas typical of the Yangtze River Delta,China(In Chinese). Acta Pedologica Sinica,2010,47(6):1110—1117
[30] Shinoda Y,Sakai Y,Uenishi H,et al. Aerobic and anaerobic toluene degradation by a newly isolated denitrifying bacterium,Thauera sp. strain DNT-1. Applied and Environmental Microbiology,2004,70 (3):1385—1392
[31] Valderrama J A,Shingler V,Carmona M,et al. AccR is a master regulator involved in carbon catabolite repression of the anaerobic catabolism of aromatic compounds in Azoarcus sp. CIB. Journal of Biological Chemistry,2014,289(4):1892—1904
[32] Boll M,Löffler C,Morris B E L,et al. Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters:Organisms,strategies and key enzymes. Environmental Microbiology,2014, 16(3):612—627
[33] Mechichi T,Stackebrandt E,Gad’on N,et al. Phylogenetic and metabolic diversity of bacteria degrading aromatic compounds under denitrifying conditions,and description of Thauera phenylacetica sp. nov.,Thauera aminoaromatica sp. nov.,and Azoarcus buckelii sp. nov. Archives of Microbiology,2002,178(1):26—35
[34] Kunapuli U,Jahn M K,Lueders T,et al. Desulfitobacterium aromaticivorans sp. nov. and Geobacter toluenoxydans sp. nov.,iron-reducing bacteria capable of anaerobic degradation of monoaromatic hydrocarbons. International Journal of Systematic and Evolutionary Microbiology,2010,60(3):686—695
[35] Sarkar J,Kazy S K,Gupta A,et al. Biostimulation of indigenous microbial community for bioremediation of petroleum refinery sludge. Frontiers in Microbiology,2016,7,DOI:10.3389/fmicb.2016.01407
Genetic Diversity of Hydrocarbons Degrading Microbial Functional Gene (bssA)in the Farmland Soil Typical of Jianghan Oilfield
ZUO Xiaohu WANG Mingxia YAO Yanhong LI Zhenlun ZHOU Zhifeng†
(College of Resources and Environment,Southwest University,Chongqing 400716,China)
Farmland soils in oil fields are liable to get polluted with petroleum hydrocarbons(PHs),which consequently leads to enrichment of some special PHs degrading microbial groups in the soils. In this study,soil samples were collected from typical farmland fields near five different oil wells in the Jianghan Oilfield located in Qianjiang City,Hubei Province for analysis of PHs degrading microbial groups,using bssA (benzylsuccinate synthase gene),a functional gene key to anaerobic benzenes degradation as biomarker,and community structure of the PHs degrading microbial groups was determined with T-RFLP(Terminalrestriction fragment length polymorphism)and clone library. On such a basis,environmental factors affecting community composition of the soil bssA were discussed. Results show that the content of polycyclic aromatic hydrocarbons(PAHs)in the soil varied in the range of 0.21~2.01 mg kg-1,which indicates that the soil in this region was not seriously contaminated with PHs. Meanwhile,the T-RFLP analysis indicates that bssA diversity varied significantly from soil sample to soil sample in this oilfield,being the highest in the soil sample the highest in PAHs content,and its dominant group was in a fairly close kinship with sulfatereducing bacterium or Geobacter. Furthermore,RDA(Redundancy analysis)reveals that the contents of soil available nitrogen,phosphorus and PAHs were all main factors affecting soil bssA diversity. To sum up,all the findings indicate that the bssA-bearing microbial groups in the soil of this oilfield are Beta-proteobacteria and Deta-proteobacteria,and in close kinships with Thauera,Azoarcus and Geobacter in phylogenesis. All these microbes might degrade PAHs through reducing metabolic processes of nitrate,sulfate,and iron.
Soil;Anaerobic degradation;Benzylsuccinate synthase gene(bssA);Community structure
S154.1
A
(责任编辑:卢 萍)
* 国家自然科学基金项目(41371477)和中央高校专项基金项目(XDJK2014B047)资助 Supported by the National Natural Science Foundation of China(No. 41371477)and the Fundamental Research Funds for the Central Universities(No. XDJK2014B047)
† 通讯作者 Corresponding author,E-mail:zhouzhf@swu.edu.cn
左小虎(1990—),男,湖北汉川人,硕士研究生,主要从事土壤微生物学研究。E-mail:375332084@qq.com
2016-11-04;
2017-01-10;优先数字出版日期(www.cnki.net):2017-02-28
10.11766/trxb201611040324
猜你喜欢
云南化工(2021年5期)2021-12-21 07:41:16
猪业科学(2021年3期)2021-05-21 02:05:36
幽默大师(2020年10期)2020-11-10 09:07:22
四川冶金(2019年5期)2019-12-23 09:04:48
中华诗词(2019年1期)2019-11-14 23:33:56
江苏农业科学(2019年5期)2019-09-02 14:01:46
经济技术协作信息(2018年30期)2018-11-22 06:21:16
猪业科学(2018年4期)2018-05-19 02:04:31
广东农业科学(2017年5期)2017-08-29 10:37:31
中国资源综合利用(2016年1期)2016-02-03 02:55:12
