
纤维素催化转化制备C5/C6烷烃燃料的反应与催化体系的研究进展
2017-06-05 15:22陈伦刚张兴华王铁军马隆龙
林产化学与工业 2017年2期
陈伦刚, 刘 勇,2, 张兴华, 张 琦, 王铁军, 马隆龙*
(1.中国科学院 广州能源研究所;中国科学院 可再生能源重点实验室;广东省新能源和可再生能源重点实验室,广东 广州 510640;2.中国科学院大学,北京 100049 )
纤维素催化转化制备C5/C6烷烃燃料的反应与催化体系的研究进展

CHEN Lungang
陈伦刚1, 刘 勇1,2, 张兴华1, 张 琦1, 王铁军1, 马隆龙1*
(1.中国科学院 广州能源研究所;中国科学院 可再生能源重点实验室;广东省新能源和可再生能源重点实验室,广东 广州 510640;2.中国科学院大学,北京 100049 )
介绍了近年来纤维素催化转化制取C5/C6烷烃的反应和催化体系的研究进展,主要论述了纤维素通过水解-加氢脱氧的一锅法过程和纤维素经C6平台化合物的加氢脱氧过程,对天然木质纤维原料、纤维素、葡萄糖及山梨醇转化为烷烃的反应路径及相应的催化剂进行了总结。反应路径主要有山梨醇、异山梨醇、HMF和己内酯反应路径,催化剂主要为金属-酸多功能催化剂,酸催化剂包括金属氧化物、分子筛、杂多酸、离子液态酸性溶剂及无机酸等;金属催化剂主要有Pd、Pt、Ru、Ir、Ni等。其中金属Ru在酸性水热环境中具有良好的催化活性,研究最为广泛。通过分析各种反应途径及相应的催化剂,提出了该研究领域面临的主要问题,并从技术角度对未来应用前景进行了展望。
纤维素;催化;烷烃;水解;加氢脱氧
人类社会发展中,能源的推动力占据着不可撼动和日趋重要的地位。根据国际能源署2013年发布的《world energy investment outlook》[1],全球对能源的投资逐年增加,其中为消费者提供能源这一项投资在2013年就达到了16 000亿美元。预计到2035年,每年供应世界能源需求的投资会逐步上升到20 000亿美元。其中,低碳能源的地位在不断凸显,持续的低碳政策信号不断强调着可再生能源的重要性,预计到2035年低碳能源供应投资会升至近9 000亿美元。这些数据表明,能源尤其是低碳能源在现代社会发展中具有重要的地位。生物质能作为可再生的碳基能源,具有来源丰富、可利用性强及可再生性3个基本特征。在全球变暖、CO2排放量居高不下的背景下,可再生碳基能源极大地刺激了全球市场对生物质的关注。生物质产业发展的一条最基本的路线是将各组分分离,通过水解或发酵等方法得到一些中间产物(如糖类等),进而将其转化为平台化合物,最后这些平台化合物经过化学或生物方法转化为所需的化学品或燃料[2-4],由生物质转化为平台化合物的过程在逐步实现商业化生产。同时,由原料一步转化为平台化合物或者高附加值化学品的过程具有操作单元少、能耗少等优势,研究潜力巨大,具有广阔的市场应用前景。例如,生物质中纤维素和半纤维素为C6和C5的糖类聚合物,它们可以经过水解-加氢脱氧过程直接转化为高附加值的烷烃(如戊烷、己烷等),避免了多步过程。同时,小分子烷烃如戊烷和己烷由于沸点较低、易分离可避免高能耗的蒸馏过程。生物质及其衍生物的氧含量很高,要生成烷烃类化合物,加氢脱氧的转化路径及催化反应体系的选择显得尤为重要。生物质及其衍生物中含氧官能团较多,例如,羟基、羧基等,亲水性较强,在水相体系中原料易分散可使反应物和催化剂充分接触。虽然水相反应具有溶剂来源广泛,成本相对低廉的特点,但由于酸水解是纤维素降解所必需的一个途径,要实现生物质催化转化为烷烃燃料,适合水相反应的耐酸、耐水热的催化剂和反应体系的开发是研究的核心之一。纤维素的水解加氢反应使用的催化剂主要为金属-酸催化体系[5-7]。水解断裂糖苷键反应中,使用的酸有杂多酸(heteropoly acid)[8-10]、离子液态酸性溶剂[11]、高温下临界水溶液[12]、改性分子筛及无机酸[13-14]等。对于加氢反应,金属是活性中心,主要有Ru[15-16]、Pt[17]等贵金属,对于非贵金属,Ni[18]是研究的热点之一。贵金属中Ru的价格相对较低,且在酸性水热环境中具有良好的加氢活性,使其得到了极为广泛的关注与研究[19-22]。笔者总结了纤维素直接转化为烷烃的反应,及其下游糖类衍生物、平台化合物的加氢脱氧反应的研究现状,并对催化剂及催化作用机制、反应途径和反应机理进行了阐述,旨在为纤维素催化转化制备烷烃燃料提供参考。
1 纤维素平台化合物转化为烃类燃料
1.1 纤维素水解降解产物转化为己烷
纤维素降解产物中葡萄糖、脱氧葡萄糖及山梨醇等化合物转化的研究热点之一,就是通过催化加氢脱氧来生产烃类燃料,如己烷、戊烷等烷烃。纤维素水相催化转化为己烷的反应中,纤维素不可避免地要经历两个主要过程,首先纤维素发生水解生成葡萄糖或其他低聚物[23-29],接着葡萄糖在金属催化剂上加氢生成C6糖醇(山梨醇和甘露醇等)[8, 30-32]。因此,分析总结纤维素水解-加氢产物的加氢脱氧过程有助于深入认识烷烃生成的反应路径和机理。
多羟基化合物和其它含氧化合物主要是以脱水-加氢、氢解及脱羧/脱羰(COx)反应来实现脱氧(图1)。对于金属-酸催化剂,发生这些反应的同时不可避免要涉及到碳链断裂的副反应,如C—C键断裂生成分子更小的烷烃如甲烷、乙烷等。一般,多羟基化合物的脱水方式有两种:一种为邻位脱水,即羟基(—OH)和邻位碳上的氢(—H)脱去一分子水生成烯烃;另一种为环脱水,即羟基和相邻或不相邻位上的羟基脱去一分子水生成环醚产物,考虑到环张力,一般环脱水产物为五元环或六元环的环醚。氢解反应主要是指金属活性位上发生C—O键断裂,较少经过酸催化的脱水过程。脱COx主要是羧酸脱去一分子CO2或羰基化合物脱去一分子CO。因此,要实现选择性地脱除氧而不破坏C—C键是比较困难的,C1~C4小分子烷烃的生成难以避免。此外,糖类的逆羟醛缩合反应也导致C—C键断裂,造成碳链破坏和C6烷烃产率的降低。例如,纤维素水解生成的葡萄糖可以发生逆羟醛缩合反应生成丙二醇、乙二醇[33-35]。

图1 水相中纤维素的反应途径
1.2 水相重整山梨醇生成小分子液态烃
小分子液态烃主要是可以用作交通燃料添加剂的C5、C6烷烃。尽管纤维素生成山梨醇的过程中会生成其他C6糖醇(甘露醇和赤藓糖醇),但这些C6糖醇经加氢脱氧反应也可转化为C5、C6烷烃。因此,山梨醇常用作加氢脱氧反应的模型反应物,通常以水作溶剂在间歇或连续反应器上考察催化剂催化山梨醇生成戊烷和己烷的催化性能,催化剂主要为酸性载体负载的贵金属Pt、Pd、Re等催化剂。
在2004年,Wisconsins大学的Dumesic团队首次提出了山梨醇水相重整制取烷烃的过程[36-37],考察了SiO2-Al2O3(SiAl)或Al2O3载体负载Pt或Pd催化剂,发现Pt催化剂可以降低山梨醇C—C键断裂的几率,比Pd催化剂低一个数量级。对于Pd-SiAl催化剂,氢气压力的升高可以减少气相产物中CO2的含量,认为山梨醇在Pd-SiAl催化剂上C—C键的断裂活性要低于金属Pd活性位上的加氢活性及酸性位上的脱水活性。此外,发现盐酸的引入有助于长链烷烃的生成,当pH值从7减到2时,气相中C5、C6烷烃的选择性逐渐增加。对于不同载体负载的Pt催化剂,Pt/ZrP和Pt/SiAl催化性能的差别主要是由载体性质造成的,例如,载体酸性是一个关键因素。与Pt/SiAl催化剂相比, Pt/ZrP催化剂可以得到更高的C5和C6烷烃选择性,说明ZrP 载体负载的Pt催化剂具有更高的C—O键断裂速率。另外,反应物浓度也会影响产物选择性,当反应物的浓度增加时,气体产物中CO2的浓度会随山梨醇转化率的降低而增加,在水相中脱水副产物1,4-脱水山梨糖醇及异山梨醇的含量也大大增加,其选择性可达99%以上。

对于山梨醇APHDO,水相反应中负载金属催化剂的稳定性直接影响到催化剂的效果。对于Pt/SiAl 催化的山梨醇APHDO,研究表明Pt/SiAl催化剂在水蒸气及水溶液中会出现Pt粒子的烧结现象,水热条件下会导致载体的比表面积下降[39]。当采用蒸汽预先老化,Pt/SiAl催化剂可以抑制水热环境中的结焦现象及载体织构性质的变化,但是预处理并不能导致产物选择性的改变。2014年,Cabiac团队制备了3种含WOx的固体酸(ZrO2-WOx, Al2O3-WOx, TiO2-WOx) 并混合Pt/ZrO2催化剂来考察山梨醇APHDO[40]。发现ZrO2与WOx的强相互作用并不能产生较强的酸性位,相反TiO2与WOx的作用不强却产生了较强的酸性位。通常在水相中酸催化的活性主要依赖于强酸性位, C5、C6烷烃的产率主要依赖于固体酸的酸性,所以TiO2-WOx催化剂的加入使得C5、C6烷烃的产率较高。
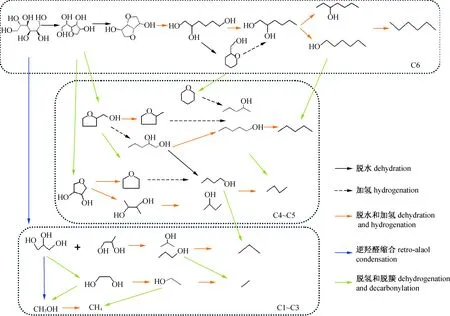
图2 山梨醇加氢脱氧的主要反应路径[38]
Vilcocq等[41]还研究了TiO2-WOx固体酸混合不同的加氢催化剂M/ZrO2(M为Pt、Ir、Pd)来考察它们对山梨醇APHDO的影响。ZrO2被选作水相催化反应中的催化剂载体,因为ZrO2是一种具有良好水热稳定性的金属氧化物,ZrO2负载金属催化剂的稳定性一般要高于SiO2-Al2O3负载催化剂,但是ZrO2的表面酸性较弱不具有较强的脱水活性。对于金属活性位,Pd和Ir金属对C5和C6烷烃的选择性较高,对反应底物具有较低的C—C键断裂活性,但由于山梨醇APHDO的转化频率(TOF)低而导致C5、C6烃的产率非常低,这与有关多元醇的研究结果相符合[42-43]。当Pt/ZrO2与酸催化剂TiO2-WOx按质量比20 ∶80混合时,催化性能具有较高的C6烃选择性和较高的反应活性,但是烃类化合物的产率仍然受到酮类和含氧杂环化合物加氢反应的影响。Cabiac团队还以反应过程中检测到的C6中间产物来分析山梨醇的APHDO反应历程[44]。C6中间产物分别为单氧化合物1-己醇、2-己醇、2,5-二甲基四氢呋喃以及2-己酮;还包括双氧中间产物1,2-己二醇和三氧化合物1,2,6-己三醇。研究指出,二级醇的脱水-加氢反应较易进行,如2-己醇容易发生脱水-加氢反应转化为正己烷。对于一级醇1-己醇的转化,反应产物为正己烷、正戊烷及CO2,主要的成分为正戊烷,说明该过程主要为脱氢-脱羧反应,而并非理想的脱水-加氢反应。然而,2-己酮和2,5-二甲基四氢呋喃的转化速率相对较低,但它们均与二级醇2-己醇的产物选择性相一致。1,2-己二醇可以依次经过两个羟基的脱水-加氢反应最后生成己烷,或是通过一次脱氢-脱羧反应得到戊烷,或连续通过两次脱氢-脱羧反应得到丁烷。在这些模型化合物的转化中,它们生成己烷的选择性均较低,而CO2的选择性较高。其中1,2,6-己三醇的反应相对更为复杂,但生成的产物结果与1,2-己二醇类似。因此,在山梨醇APHDO过程中应当尽量减少脱氢反应的发生或避免脱羧反应,尽量促进加氢反应及脱水反应,从而提高己烷的产率。
另外,马隆龙团队研究了分子筛负载Ni催化剂催化山梨醇APHDO生成汽油组分烷烃的反应,发现Ni/HZSM-5(n(Si)/n(Al)=38)具有良好的催化效果[45-46]。该催化剂的煅烧温度对其物化性质有较大影响,从而影响反应产物的生成。在773 K煅烧温度下,Ni/HZSM-5催化剂具有较高的比表面积,载体与Ni物质具有较强的相互作用,催化剂表面上的NiO可以被完全还原,获得的烷烃产率最高达47.6%,烷烃中有76.4%为C5、C6烷烃。此外,还发现HZSM-5载体被纯硅MCM- 41分子筛修饰后可进一步显著提高液态烷烃的产率[47]。当催化剂载体含40%的MCM- 41时,C5、C6烷烃的总选择性可达到98.7%(山梨醇的转化率为67.1%)。研究指出优化催化剂表面金属-酸性质可以实现山梨醇高选择性转化为液态烷烃,MCM-41的添加可以增大催化剂的比表面积及吸附能力,从而加剧山梨醇与氢气的相互作用,同时通过抑制HZSM-5骨架中Al酸性位的作用来调节催化剂表面酸性位的分布和含量[47]。该团队还以木糖醇为反应物考察了2%Ni/HZSM-5及2%Ni/MCM22催化剂在513 K,4.0 MPa条件下的催化效果,分别得到91%及95%的戊烷产率[48]。
最近,日本的Tomishige团队在水/正十二烷两相体系中,以HZSM-5为助催化剂,研究了不同催化剂对山梨醇转化为己烷的性能(己烷产率顺序):Ir-ReOx/SiO2> Rh-ReOx/SiO2> Ru/C > 骨架Ni > Pt/C[49]。Ir-ReOx/SiO2可以得到96.3%的己烷产率,当使用HI-H3PO4混合酸,可以获得C12~C18的长链烃[50]。水与HI比会影响反应结果,当水含量增加、HI含量下降时可以提高烃类化合物的选择性,但总产率会降低。当山梨醇、HI、H3PO4的物质的量比为1 ∶3 ∶8时,水为34%,反应12 h后可以得到62.07%的烃类产率。
通常水溶液环境中山梨醇分子进行加氢脱氧需要金属-酸多功能催化活性位,但在转化过程中分子结构及化学性质较稳定的异山梨醇的生成及进一步活化是影响催化剂活性和C6烷烃产率的关键[51]。异山梨醇是含双四氢呋喃环结构的分子,是山梨醇1,4-和3,6-位羟基环脱水的产物。当采用Pt/NbOPO4催化剂能有效地促进山梨糖醇APHDO生成己烷的效率并提高产率,250 ℃,4.0 MPa 氢压力下Pt/NbOPO4催化剂在保持高活性的情况下,C5/C6烷烃的产率可达60%[51]。图3表明山梨醇加氢脱氧过程涉及脱水反应、氢解和C—C键裂解反应。其中异山梨醇是主要的脱水产物,也是重要的反应中间物,它会进一步经历开环和后续的氢解反应形成己烷。研究发现Pt/NbOPO4山梨糖醇脱水和异山梨醇氢解的活化能分别为72.7和147.6 kJ/mol, 要比Pt/ZrP和Pt/H-β催化剂低得多,这可能与NbOPO4载体上具有较多的强酸位作用及NbOX对环氧C—O键断裂的促进作用有关。研究指出,异山梨醇氢解是决定整个反应过程速率的关键步骤,降低异山梨醇的活化能是提高反应活性的关键[51]。

图3 Pt/NbOPO4催化山梨醇氢解为己烷的反应路径[51]
1.3 葡萄糖低聚物、(脱氧)葡萄糖转化为液态小分子烃
山梨醇可以从葡萄糖或葡萄糖的聚合物(纤维二糖、纤维素以及淀粉等)转化而来。因此研究纤维素的单体和低聚物(纤维二糖)的转化有助于认识纤维素转化为烷烃的过程。Gagné团队以葡萄糖为底物,Et2SiH2作为氢源, [(POCOP)IrH(丙酮)]+[B-(C6F5)4]-(POCOP = 2,6-[PO(tBu)2]2C6H3)为催化剂,室温下实现葡萄糖还原为己烷 (图4)[52]。原位核磁结果表明葡萄糖的还原反应是通过逐步的脱氧过程来实现,葡萄糖C1位C—O键的氢硅烷化速率的规律是:C1 赤道位, OMe> C1 轴向位, OMe≫ C1 赤道位, OSiR3> C1 轴向位, OSiR3。除了C1上的脱氧过程非常迅速外,同位素标记13C NMR可以说明其它位置上的脱氧过程不具有选择性,NMR谱图表明它们的出现几乎和消失是同时的,这也说明该反应中的消除反应是竞争反应。此外,C1位以甲氧基为取代基的原料所得到的产物主要为2-甲基戊烷或3-甲基戊烷,而C1位以-OSiR3为取代基的原料所得到的产物主要是正己烷。

图4 铱催化葡萄糖氢硅烷化还原为己烷[52]
2014年,Gagné团队还以商业催化剂B(C6H5)3替换Ir催化剂来研究葡萄糖的转化[53],该Lewis酸通常被用作一级醇和二级醇的还原反应[54-55]。研究表明该催化剂也可以将葡萄糖转化为己烷、己烯及其异构体(图5中反应式(a))。当使用硅烷化的甲基葡萄糖苷作为反应底物时,二者的区别在于[(POCOP)IrH(丙酮)]+[B-(C6F5)4]-所得到的主要产物不是正己烷,而B(C6H5)3所得到的主要产物为正己烷(图5中反应式(b))。Ir催化剂所得的主要产物为含氧化合物,以及少量的烷烃;而B(C6H5)3所得的主要产物为烃类化合物, B(C6H5)3催化反应速率更快。此外,产物的分布也依赖于反应底物,如表1所示。以1,6-脱水葡萄糖为反应底物时可以得到更多的甲基戊烷,2-甲基戊烷和3-甲基戊烷共占71.9%;而以山梨醇为反应底物时则生成的甲基戊烷最少,仅为14.1%。产物中的甲基戊烷分布也随反应底物的不同而有差别,2-脱氧葡萄糖所得的3-甲基戊烷最高,达42.5%。当以Me2EtSi基团保护的山梨醇(Me2EtSi-6)为反应底物时,主要产物为端基位上的氧被还原的化合物。相同条件下,未进行保护的山梨醇反应后的结果相类似,即1,6-脱氧山梨醇的产率也可达到70%,这在动力学上表明伯醇比仲醇更容易还原,该结果与前人的研究相一致[54]。

图5 催化葡萄糖和甲基葡萄糖苷的脱氧反应[53]
日本东北大学的Tomishige团队使用Ir-ReOx/SiO2+HZSM-5为催化剂,在水/正十二烷(质量比4 ∶3)中将葡萄糖及纤维二糖转化为烷烃[49]。以葡萄糖为反应底物可以得到产率为98.2%的烷烃及94.4%的正己烷;以纤维二糖为反应底物可以得到产率为98.9%的烷烃及94.8%的正己烷。此外,比利时鲁汶大学的Sels团队使用Ru/C+H4SiW12O40为催化剂,在水/正十烷(质量比20 ∶15)中将葡萄糖及果糖转化为烷烃[55],以葡萄糖为反应底物,产物为44.4%的正己烷、16.5%的正戊烷及7.9%的环己烷;以果糖为反应底物,产物为35.2%的正己烷、7.3%的正戊烷及2.9%的环己烷。

表1 不同反应底物加氢脱氧生成烷烃的产物分布
2 纤维素和天然生物质原料直接转化为烷烃
2.1 纤维素的直接转化
纤维素作为高分子化合物是由葡萄糖单体构成,随着葡聚糖的聚合度不断上升,其分子中的羟基数目不断增加,分子内氢键的作用也不断加强。当聚合度达到一定程度后,所得到的化合物在水中的溶解度就会大大下降,以致于不能溶于水及传统的溶剂,这使得纤维素的转化难度增加。由于纤维素不溶于水且含氧量高、纤维素通过脱氧一步转化为烷烃较为困难。
日本的Iwamoto团队[56]考察了一系列被聚乙烯吡咯烷酮 (PVP)保护的金属催化剂,在亚临界水条件下各金属对纤维素转化为烷烃的加氢脱氧性能如下:Ru ≫ Ir > Rh、 Pd、 Pt及 Au。Ru具有较高的C—C键断裂活性及加氢活性,生成的产物主要为甲烷和一些小分子气体烷烃,而其它金属催化剂所得到的产物主要为醇类化合物。改变载体的种类可以调变金属Ru的催化性能,Al2O3、 CeO2及活性炭为载体时,金属Ru的催化性能并不改变;而TiO2、ZrO2、SiO2及SiO2-Al2O3为载体时,导致反应活性降低,同时催化性能发生巨大改变,主要产物为醇类而非烷烃类化合物。Murata等[57]研究了不同载体(HZSM-5,USY,Y,SiO2-Al2O3)负载的Pt催化剂,认为Pt/HZSM-5催化剂对纤维素氢解反应具有较好的活性,可以得到C2~C9烷烃。将纤维素用醇进行预处理后可以进一步提高烷烃的产率,在623 K下对纤维素用正己醇进行预处理后,可以得到89%的C2~C9烷烃,而只有6%的CH4或COx。当使用未预处理的纤维素为原料时,催化剂中Re的引入虽然可以增加烷烃产物中C2~C9烷烃的选择性,最高可达76.8%,但是烷烃和COx的产率下降,从89%下降到52%。Sekine等[58]研究了Pt/H-beta 催化剂,结果表明不同载体对纤维素转化率有明显影响,转化率依次为:Pt/H-FER(18) < Pt/H-MOR(90) < Pt/HZSM-5(38) < Pt/H-MOR(10.2) < Pt/H-beta(150) < Pt/H-beta(25) < Pt/H-USY(14),低Si/Al比具有大孔结构的载体所负载的Pt催化剂具有较高的纤维素氢解活性,这可能是纤维素降解而来的中间产物,如葡萄糖容易在大孔结构的分子筛内进行扩散所导致,另外Pt金属能较好的负载在分子筛内部也是重要原因之一,不过所得到的主要产物为C3~C4的烃类化合物,总产率在10%左右。
球磨通常能降低纤维素的结晶度,被认为是一种有效增加纤维素在水中溶胀或溶解度的方式[13, 59-61]。Shrotri等[61]对使用无机酸处理过的微晶纤维素进行球磨后,用Ni-Pt/Al2O3催化加氢可以得到90%的山梨醇和甘露醇。发现球磨后的纤维素可形成α(1→6)键从而大大增加了低聚糖在水中的溶解度。Hilgert等[13]同样使用球磨法处理过的纤维素,其转化为糖醇的产率可提升到94%。同样,球磨后的纤维素可以被催化转化为更高产率的己烷。Liu等[62]使用球磨的纤维素为反应物,在水/正十二烷中采用Ir-ReOx/SiO2+HZSM-5催化剂可以将纤维素转化为正己烷,产率达83%。当原料为微晶纤维素,其产率降低为78%,反应温度从463 K上升到483 K。该研究认为反应路径为高温水或HZSM-5可以将纤维素降解为水溶性的低聚糖进而水解为葡萄糖;继而葡萄糖被Ir-ReOx/SiO2催化剂加氢为山梨醇;最后山梨醇被Ir-ReOx/SiO2+HZSM-5催化氢解为正己烷(图6)。

图6 纤维素转化为正己烷的反应路径[62]
Beeck等[55]使用H4SiW12O40+htTSARu/C催化剂(H4SiW12O40预处理过的改性Ru/C催化剂)在水/正十烷两相体系中也可将纤维素直接转化为己烷和戊烷。研究认为纤维素转化为烷烃的反应路径为:纤维素水解产生葡萄糖,然后脱水生成5-羟甲基糠醛(5-HMF),生成的5-HMF迅速加氢为2,5-二羟甲基呋喃(2,5-DHMF),而2,5-DHMF在加氢或氢解作用下得到2,5-二甲基四氢呋喃(2,5-DMTHF),该化合物通过连续的开环水解及脱水加氢反应最后得到正己烷(图7)。同时HMF的羰基通过加氢或氢解反应也会产生一系列的呋喃类化合物,进而通过开环反应得到2,5-己二酮,最终加氢脱氧也可以得到正己烷。此外,2,5-DHMF的开环反应得到的一级醇正己醇也是产生己烷和戊烷的途径。该反应有3个关键步骤:1)双相体系的使用可以使活性中间物在有机相进行更好的反应;2)控制加热速率使反应逐步释放葡萄糖及形成HMF,从而避免副反应的发生;3)Ru/C催化剂的改性可避免催化葡萄糖加氢生成山梨醇从而促进HMF的生成。

图7 纤维素转化为烷烃的HMF反应路径[55]
国内,马隆龙团队开展了纯水相体系中纤维素转化为己烷的研究,采用无机酸或固体酸与Ru/C组合的催化剂,考察了MCM-41, HZSM-5, SBA-15,γ-Al2O3或层状化合物与Ru/C-磷酸组合催化剂的反应效果[63]。结果表明Ru/C-层状化合物-磷酸的效果最优,最高获得72%的己烷产率,且催化剂具有良好的循环使用性能,循环使用5次后仍可获得52.8%的己烷产率,使用过的LiNbMoO6经焙烧后可恢复催化活性。Ru/C-LiNbMoO6-磷酸对不同底物的催化效果如下(己烷产率):纤维素>葡萄糖>山梨醇>异山梨醇。相同条件下,异山梨醇的转化率仅为14.1%,难以转化为己烷。研究认为层状催化剂具有较大孔径及层状结构的特征对反应影响显著,XRD实验表明葡萄糖和山梨醇可以进入层间孔道,而异山梨醇无法进入,由于层状催化剂层间孔道的空间位阻作用,进入的葡萄糖和山梨醇转化为异山梨醇的过程被抑制,使反应朝有利于己烷生成的方向进行。提出以下反应路径:在磷酸及高温水的催化作用下,纤维素逐渐水解释放出可溶性的低聚糖及葡萄糖,而葡萄糖被层状化合物吸附进入层间孔道,同时经Ru/C加氢葡萄糖转化为山梨醇,逐步释放的葡萄糖及山梨醇可以有效地进入层状化合物的层间孔道,由于孔道空间位阻作用山梨醇难以发生环脱水反应生成稳定的异山梨醇,促进了山梨醇通过连续的C—O键断裂反应生成己烷。
2.2 天然木质纤维原料的直接转化
木质纤维原料主要包括纤维素、半纤维素及木质素3种组分。纤维素和半纤维素分别由C6和C5的糖单元聚合而成,木质素则是由3种不同的苯丙烯单体交联聚合而成。除此之外,天然原料还含有灰分、不纯无机物和有机物等杂质,所以天然木质纤维原料的转化要比纤维素复杂得多。近年来,从应用角度出发,木质纤维原料不经化学预处理或分离,直接在温和的条件下实现加氢脱氧生成液态烷烃的研究开始受到关注。
马隆龙团队研究了几种木质纤维原料的直接转化,例如,松木屑、玉米秸秆、玉米芯、麦秆和稻秆(图8)[64]。以松木屑为底物,考察了3种层状催化剂LiNbMoO6、LiNbWO6及LiTaMoO6的反应效果,发现Ru/C-LiTaMoO6-磷酸组合的催化剂可以高效地将松木屑转化为小分子液态烷烃如己烷和戊烷。以玉米秸秆为原料,依据纤维素和半纤维素的碳含量,可以得到82.4%的小分子液态烃,即60.4%的C6烃和22%的C5烃。以玉米芯为原料可以得到51.4%的木质素降解产率,产物主要为愈创木酚类、单酚类、烃类、醇类等化合物。

图8 三组分催化剂一锅法转化天然木质纤维原料生成的产物[64]
马隆龙团队提出了天然木质纤维原料的反应途径(图9)[64],主要有3种路径。发现转化过程中主要的C6中间产物为正己醇、山梨醇、脱水山梨糖醇、异山梨醇、甲基环戊酮、己酸、γ-己内酯、2-羟甲基四氢吡喃、5-羟甲基糠醛及2-甲基戊酸。分析表明:木质纤维原料转化为烷烃的过程中,除了中间产物山梨醇的反应路径和中间产物5-羟甲基糠醛的反应路径外,还可能存在另一种反应路径,即中间产物内酯类的反应路径,发现这些内酯(γ-己内酯、己酸及2-甲基戊酸)也可以有效地转化为戊烷和己烷。使用相同的催化剂,以γ-己内酯、己酸、2-甲基戊酸为反应底物可以分别得到气相选择性为97.8%、96.8%、97.6%的液态烷烃,降低来源于γ-己内酯的己酸的量可以得到83.3%的己烷产率。相比其它3种木质纤维原料,稻秆和麦秆的小分子液态烃的产率相对较低,不到42%。分析认为,其主要原因可能是由于它们含有其它原料10倍左右的硫元素,这些硫元素以有机物形式存在导致金属催化剂中毒、活性严重下降,同时发现氨基酸中的硫(含量相当于稻秆或麦秆中的硫)对金属催化剂的催化性能影响明显,使催化剂的加氢活性显著下降,造成大量未加氢完全的不饱和气体产物生成。然而,对于灰分中无机硫的存在却没有这样的影响,对金属催化剂性能的影响较小。
此外,华东理工大学王艳芹团队近期报道了天然木质生物质(软木:松树、白松、落叶松、杉树,硬木:樟树、白桦树、白杨树)催化转化生成液态烃燃料的工作[65]。该研究构建了多功能Pt/NbOPO4催化剂,木质生物质不经化学预处理或分离在温和的条件下直接加氢脱氧生成液态烷烃(纤维素、半纤维素和木质素组分分别转化为己烷、戊烷和烷基环己烷),质量收率高达28.1%。Pt/NbOPO4催化剂中NbOX物种起到了决定性的作用,既作为酸中心发挥水解、脱水的作用,同时又具有活化C—O键的能力,促进了加氢脱氧过程的进行,DFT理论计算和原位非弹性中子散射实验进一步证实了这一结果。

图9 木质纤维原料的反应网络图[64]
3 展 望
生物质催化转化合成C5/C6液态烃燃料是近10年迅速发展起来的新技术,目前已成为国内外科研人员的研究热点。无论是通过生物质原料水解-加氢脱氧一锅法,还是经糖平台化合物的加氢脱氧法,制备生物质液态烃燃料仍存在各种问题与挑战。目前报道较多的研究主要是以模型化合物(纤维素和山梨醇等)为原料的转化,研究集中在催化剂和相应的反应路径及其机理,也有少量以天然生物质为原料的研究。这些研究说明,此项技术从路线上来说是可行的,但实际应用仍需长时间研究与开发。
需要解决的关键问题归纳起来有两个方面,即催化剂的耐用性和生产效率。催化剂的耐用性指的是在复杂多相体系中催化剂保持高活性和高产率的稳定性,以及导致催化剂失活的复杂因素,这需要从催化剂及反应体系的研究来解决。而影响生产效率的因素涉及到多方面,例如,原料本身就是结构稳定的高分子材料,能量密度低、含氧量高;木质纤维原料仍不能高效转化为平台化合物,副产物多,目标平台化合物收率低;含氧平台化合物的脱氧效率及液态烃的产率低。如果能够充分利用木质纤维原料中3种组分在转化过程中的副产物,则能够有效提高转化过程中生物质的碳利用率,从而提高技术经济性。目前加氢脱氧反应均涉及到高温、高压条件,以及普遍使用贵金属催化剂,大大提高了成本,阻碍了该技术的发展与应用。因此,进一步降低反应条件,寻找可替代的高效非贵金属催化剂将是实现技术应用的前提。其次是水解和水相加氢脱氧技术中涉及到的反应过程对反应设备和工程工艺的要求苛刻,尤其进行大规模生产在反应设备和工程工艺上均无经验可参考,有可能会面临不确定的困难和问题。同时,由于木质纤维原料的低密度特点决定了转化技术的规模化在空间和时间上均有所限制,即实现大规模生产需要更大或更多的反应设备,有可能导致生产效率偏低。
此外,氢气的需求也制约技术的发展和应用。加氢脱氧技术是去除生物质原料中氧元素的重要手段,由于生物质原料中氧含量高,脱氧过程中要消耗大量的氢气,因此解决大量廉价氢气的来源直接关系到技术经济性问题。这也是广大研究者为实现地球上分布最广泛、最多的生物质资源的转化利用而思考的问题。尽管面临着诸多困难与挑战,生物质催化转化制备烃类燃料技术的进一步发展和成熟将有助于液态烃燃料的生产从生物质原料的种类及供应系统、技术革新及集成、规模化应用等方面形成一种新的发展格局,为人类利用生物质资源提供新的思路。
[1] IEA. World Energy Investment Outlook 2014[M]. International Energy Agency.
[2] ALONSO D M, WETTSTEIN S G, DUMESIC J A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals [J]. Chemical Society Reviews, 2012,41(24): 8075-8098.
[3] CORMA A, IBORRA S, VELTY A. Chemical routes for the transformation of biomass into chemicals [J]. Chemical Reviews, 2007, 107(6): 2411-2502.
[4] GRAY H L, STAUD C J. Recent advances in cellulose and starch chemistry [J]. Chemical Reviews, 1928, 4(4): 355-373.
[5] CLIMENT M J, CORMA A, IBORRA S. Heterogeneous catalysts for the one-pot synthesis of chemicals and fine chemicals [J]. Chemical Reviews, 2011, 111(2): 1072-1133.
[6]FUKUOKA A, DHEPE P L. Catalytic conversion of cellulose into sugar alcohols [J]. Angewandte Chemie, 2006, 118(31): 5285-5287.
[7] HUBER G W, IBORRA S, CORMA A. Synthesis of transportation fuels from biomass:Chemistry, catalysts, and engineering [J]. Chemical Reviews, 2006, 106(9): 4044-4098.
[8] NEGAHDAR L, OLTMANNS J U, PALKOVITS S, et al. Kinetic investigation of the catalytic conversion of cellobiose to sorbitol [J]. Applied Catalysis B: Environmental, 2014, 147: 677-683.
[9] OGASAWARA Y, ITAGAKI S, YAMAGUCHI K, et al. Saccharification of natural lignocellulose biomass and polysaccharides by highly negatively charged heteropolyacids in concentrated aqueous solution [J].ChemSusChem, 2011, 4(4): 519-525.
[10] PALKOVITS R, TAJVIDI K, RUPPERT A M, et al.Heteropoly acids as efficient acid catalysts in the one-step conversion of cellulose to sugar alcohols [J]. Chemical Communications, 2011, 47(1): 576-578.
[11] ZHU Y H, KONG Z N, STUBBS L P, et al.Conversion of cellulose to hexitols catalyzed by ionic liquid-stabilized ruthenium nanoparticles and a reversible binding agent [J]. ChemSusChem, 2010, 3(1): 67-70.
[12] LUO C, WANG S, LIU H C. Cellulose conversion into polyols catalyzed by reversibly formed acids and supported ruthenium clusters in hot water [J].Angewandte Chemie, 2007, 119(40): 7780-7783.
[13] HILGERT J, MEINE N, RINALDI R, et al.Mechanocatalytic depolymerization of cellulose combined with hydrogenolysis as a highly efficient pathway to sugar alcohols [J]. Energy & Environmental Science, 2013, 6(1): 92-96.
[14] PHILIPPAERTS A, GOOSSENS S, VERMANDEL W, et al. Design of Ru-zeolites for hydrogen-free production of conjugated linoleic acids [J]. ChemSusChem, 2011, 4(6): 757-767.
[15] KOBAYASHI H, MATSUHASHI H, KOMANOYA T, et al. Transfer hydrogenation of cellulose to sugar alcohols over supported ruthenium catalysts [J]. Chemical Communications, 2011, 47(8): 2366-2368.
[16] OP DE BEECK B, GEBOERS J, VAN DE VYVER S, et al. Conversion of (ligno)cellulose feeds to isosorbide with heteropoly acids and ru on carbon [J]. ChemSusChem, 2013, 6(1): 199-208.
[17] PARK D S, YUN D, KIM T Y, et al. A mesoporous carbon-supported Pt nanocatalyst for the conversion of lignocellulose to sugar alcohols [J]. ChemSusChem, 2013, 6(12):2281-2289.
[18] DING L N, WANG A Q, ZHENG M Y, et al. Selective transformation of cellulose into sorbitol by using a bifunctional nickel phosphide catalyst [J].ChemSusChem, 2010, 3(7): 818-821.
[19] HUANG Y B, DAI J J, DENG X J, et al.Ruthenium-catalyzed conversion of levulinic acid to pyrrolidines by reductive amination [J]. ChemSusChem, 2011, 4(11): 1578-1581.
[20] MIYAZAWA T, KOSO S, KUNIMORI K, et al. Development of a Ru/C catalyst for glycerol hydrogenolysis in combination with an ion-exchange resin [J]. Applied Catalysis A:General, 2007, 318: 244-251.
[21] OLIVIERO L, BARBIER-JR J, DUPREZ D, et al. Catalytic wet air oxidation of phenol and acrylic acid over Ru/C and Ru-CeO2/C catalysts [J]. Applied Catalysis B:Environmental, 2000, 25(4): 267-275.
[22] ROSSETTI I, FORNI L. Effect of Ru loading and of Ru precursor in Ru/C catalysts for ammonia synthesis [J]. Applied Catalysis A:General, 2005, 282(1/2): 315-320.
[23] FANG Z, ZHANG F, ZENG H Y, et al. Production of glucose by hydrolysis of cellulose at 423 K in the presence of activated hydrotalcite nanoparticles [J]. Bioresource Technology, 2011, 102(17): 8017-8021.
[24] KOBAYASHI H, YABUSHITA M, KOMANOYA T, et al. High-yielding one-pot synthesis of glucose from cellulose using simple activated carbons and trace hydrochloric acid [J].ACS Catalysis, 2013, 3(4): 581-587.
[25] LAI D M, DENG L, LI J A, et al.Hydrolysis of cellulose into glucose by magnetic solid acid [J]. ChemSusChem, 2011, 4(1): 55-58.
[26] LANZAFAME P, TEMI D M, PERATHONER S, et al. Direct conversion of cellulose to glucose and valuable intermediates in mild reaction conditions over solid acid catalysts [J]. Catalysis Today, 2012, 179(1): 178-184.
[27] PANG J F, WANG A Q, ZHENG M Y, et al. Hydrolysis of cellulose into glucose over carbons sulfonated at elevated temperatures [J]. Chemical Communications, 2010, 46(37): 6935-6937.
[28] TAKAGAKI A, TAGUSAGAWA C, DOMEN K. Glucose production from saccharides using layered transition metal oxide and exfoliated nanosheets as a water-tolerant solid acid catalyst [J]. Chemical Communications, 2008 (42): 5363-5365.
[29] WANG H Y, ZHANG C B, HE H, et al. Glucose production from hydrolysis of cellulose over a novel silica catalyst under hydrothermal conditions [J]. Journal of Environmental Sciences, 2012, 24(3): 473-478.
[30] DENG W P, LIU M, TAN X S, et al.Conversion of cellobiose into sorbitol in neutral water medium over carbon nanotube-supported ruthenium catalysts [J]. Journal of Catalysis, 2010, 271(1): 22-32.
[31] LI J R, SOARES H S M P, MOULIJN J A, et al.Simultaneous hydrolysis and hydrogenation of cellobiose to sorbitol in molten salt hydrate media [J]. Catalysis Science & Technology, 2013, 3(6): 1565-1572.
[32] WANG D, NIU W Q, TAN M H, et al.Pt nanocatalysts supported on reduced graphene oxide for selective conversion of cellulose or cellobiose to sorbitol [J]. ChemSusChem, 2014, 7(5): 1398-1406.
[33] PANG J F, ZHENG M Y, WANG A Q, et al. Catalytic hydrogenation of corn stalk to ethylene glycol and 1,2-propylene glycol [J]. Industrial & Engineering Chemistry Research, 2011, 50(11): 6601-6608.
[34] TAI Z J, ZHANG J Y, WANG A Q, et al.Catalytic conversion of cellulose to ethylene glycol over a low-cost binary catalyst of Raney Ni and tungstic acid [J]. ChemSusChem, 2013, 6(4): 652-658.
[35] ZHENG M Y, WANG A Q, JI N, et al.Transition metal-tungsten bimetallic catalysts for the conversion of cellulose into ethylene glycol [J]. ChemSusChem, 2010, 3(1): 63-66.
[36] HUBER G W, CORTRIGHT R D, DUMESIC J A. Renewable alkanes by aqueous-phase reforming of biomass-derived oxygenates [J]. Angewandte Chemie, 2004.116(12), 1575-1577.
[37] LI N, TOMPSETT G A, HUBER G W. Renewable high-octane gasoline by aqueous-phase hydrodeoxygenation of C5and C6carbohydrates over Pt/Zirconium phosphate catalysts [J]. ChemSusChem, 2010, 3(10): 1154-1157.
[38] LI N, HUBER G W. Aqueous-phase hydrodeoxygenation of sorbitol with Pt/SiO2-Al2O3: Identification of reaction intermediates [J]. Journal of Catalysis, 2010, 270(1): 48-59.
[39] VILCOCQ L, CABIAC A, ESPECEL C, et al.Study of the stability of Pt/SiO2-Al2O3catalysts in aqueous medium: Application for sorbitol transformation [J]. Catalysis Communications, 2011, 15(1): 18-22.
[40] VILCOCQ L, KOERIN R, CABIAC A, et al. New bifunctional catalytic systems for sorbitol transformation into biofuels [J]. Applied Catalysis B:Environmental, 2014, 148: 499-508.
[41] VILCOCQ L, CABIAC A, ESPECEL C, et al. Hydrocarbon fuel synthesis from sorbitol over bifunctional catalysts: Association of tungstated titania with platinum, palladium or iridium [J].Catalysis Today, 2015, 242: 91-100.
[42] AMADA Y, SHINMI Y, KOSO S, et al.Reaction mechanism of the glycerol hydrogenolysis to 1,3-propanediol over Ir-ReOx/SiO2catalyst [J]. Applied Catalysis B:Environmental, 2011, 105(1/2): 117-127.
[43] DAVDA R R, SHABAKER J W, HUBER G W, et al.Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts [J]. Applied Catalysis B:Environmental, 2003, 43(1): 13-26.
[44] VILCOCQ L, CABIAC A, ESPECEL C, et al.New insights into the mechanism of sorbitol transformation over an original bifunctional catalytic system [J]. Journal of Catalysis, 2014, 320: 16-25.
[45] ZHANG Q, WANG T J, LI B, et al.Aqueous phase reforming of sorbitol to bio-gasoline over Ni/HZSM-5 catalysts [J]. Applied Energy, 2012, 97: 509-513.
[46] ZHANG Q, WANG T J, XU Y, et al.Production of liquid alkanes by controlling reactivity of sorbitol hydrogenation with a Ni/HZSM-5 catalyst in water [J]. Energy Conversion and Management, 2014, 77: 262-268.
[47] ZHANG Q, JIANG T, LI B, et al. Highly selective sorbitol hydrogenolysis to liquid alkanes over Ni/HZSM-5 catalysts modified with pure silica MCM-41 [J].ChemCatChem, 2012, 4(8): 1084-1087.
[48] JIANG T, ZHANG Q, WANG T J, et al.High yield of pentane production by aqueous-phase reforming of xylitol over Ni/HZSM-5 and Ni/MCM22 catalysts [J]. Energy Conversion and Management, 2012, 59: 58-65.
[49] CHEN K Y, TAMURA M, YUAN Z L, et al. One-pot conversion of sugar and sugar polyols ton-alkanes without C-C Dissociation over the Ir-ReOx/SiO2catalyst combined with H-ZSM-5 [J]. ChemSusChem, 2013,6(4): 613-21.
[50] LV D C, LIU Y Q, ZHANG B B, et al. Production of liquid hydrocarbons from sorbitol by reduction with hydroiodic acid [J]. Energy & Fuels, 2014, 28(6): 3802-3807.
[51] XI J X, XIA Q N, SHAO Y, et al. Production of hexane from sorbitol in aqueous medium over Pt/NbOPO4catalyst [J]. Applied Catalysis B: Environmental, 2016, 181: 699-706.
[52] MCLAUGHLIN M P, ADDUCI L L, BECKER J J, et al. Iridium-catalyzed hydrosilylative reduction of glucose to hexane(s) [J]. Journal of the American Chemical Society, 2013, 135(4): 1225-1227.
[53] ADDUCI L L, MCLAUGHLIN M P, BENDER, T A, et al. Metal-free deoxygenation of carbohydrates [J].Angewandte Chemie-International Edition, 2014, 53(6): 1646-1649.
[54] GEVORGYAN V, LIU J X, RUBIN M, et al.A novel reduction of alcohols and ethers with a HSiEt3/catalytic B(C6F5)3system [J]. Tetrahedron Letters, 1999, 40(50): 8919-8922.
[55]OP DE BEECK B, DUSSELIER M, GEBOERS J, et al. Direct catalytic conversion of cellulose to liquid straight-chain alkanes [J]. Energy & Environmental Science, 2015, 8(1): 230-240.
[56] OSAKA Y, IKEDA Y, HASHIZUME D, et al.Direct hydrodeoxygenation of cellulose and xylan to lower alkanes on ruthenium catalysts in subcritical water [J]. Biomass and Bioenergy, 2013, 56: 1-7.
[57] MURATA K, LIU Y Y, INABA M, et al.Hydrocracking of biomass-derived materials into alkanes in the presence of platinum-based catalyst and hydrogen [J]. Catalysis Letters, 2010, 140(1): 8-13.
[58] KATO Y, SEKINE Y. One pot direct catalytic conversion of cellulose to hydrocarbon by decarbonation using Pt/H-beta zeolite catalyst at low temperature [J]. Catalysis Letters, 2013, 143(5): 418-423.
[59] LIAO Y H, LIU Q Y, WANG T J, et al.Zirconium phosphate combined with Ru/C as a highly efficient catalyst for the direct transformation of cellulose to C6alditols [J]. Green Chemistry, 2014, 16(6): 3305-3312.
[60] MEINE N, RINALDI R, SCHUTH F. Solvent-free catalytic depolymerization of cellulose to water-soluble oligosaccharides [J]. ChemSusChem, 2012, 5(8): 1449-1454.
[61] SHROTRI A, LAMBERT L K, TANKSALE A, et al. Mechanical depolymerisation of acidulated cellulose: Understanding the solubility of high molecular weight oligomers [J]. Green Chemistry, 2013, 15(10): 2761-2770.
[62] LIU S B, TAMURA M, NAKAGAWA Y, et al. One-pot conversion of cellulose inton-Hexane over the Ir-ReOx/SiO2catalyst combined with HZSM-5 [J]. ACS Sustainable Chemistry & Engineering, 2014, 2(7): 1819-1827.
[63] LIU Y, CHEN L G, WANG T J, et al. High yield of renewable hexanes by direct hydrolysis-hydrodeoxygenation of cellulose in aqueous phase catalytic system [J]. RSC Advances, 2015, 5: 11649-11657.
[64]LIU Y, CHEN L G, WANG T J, et al. One-pot catalytic conversion of raw lignocellulosic biomass into gasoline alkanes and chemicals over LiTaMoO6and Ru/C in aqueous phosphoric acid [J]. ACS Sustainable Chemistry & Engineering, 2015, 3(8): 1745-1755.
[65]XIA Q N, CHEN Z J, SHAO Y, et al. Direct hydrodeoxygenation of raw woody biomass into liquid alkanes [J]. Nature Communications, 2016, 7:11162.
Progress on Reaction and Catalyst for Production of C5/C6 Alkane Fuels from Cellulose by Catalytic Conversion
CHEN Lungang1, LIU Yong1,2, ZHANG Xinghua1, ZHANG Qi1, WANG Tiejun1, MA Longlong1
(1. Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences; Key Laboratory of Renewable Energy,Chinese Academy of Sciences; Guangdong Key Laboratory of New and Renewable Energy Research and Development,Guangzhou 510640, China;2. University of Chinese Academy of Sciences, Beijing 100049, China )
The reactions and catalytic systems for the production of C5/C6 alkanes from cellulose or lignocellulosic biomass were summarized. The reaction pathways mainly included one-pot conversion of cellulose to alkanes by hydrolysis-hydrodeoxygenation; and hydrocarbon fuels produced from C6 platform compounds by hydrodeoxygenation. The reaction pathways and catalysts of alkanes production from raw lignocelluloses, cellulose, glucose and sorbitol were summarized. And the reaction pathways mainly included sorbitol, isosorbide, HMF and caprolactone routes. The metal-acid multifunctional catalysts were widely used; acid catalysts included metal oxides, zeolites, heteropoly acids, ionic liquid acid and inorganic acid, etc., and the metal catalysts were mainly Pd, Pt, Ni, Ru, Ir, etc. The metal Ru was popularly studied due to its excellent catalytic performance under the acidic hydrothermal environment. Finally, for the state-of-the-art technologies in the conversion of biomass to C5/C6 alkanes, the main uncertainties, bottlenecks and research needs were concluded and prospected.
cellulose; catalysis; alkane; hydrolysis; hydrodeoxygenation
10.3969/j.issn.0253-2417.2017.02.003
2016- 07-20
国家自然科学基金青年项目(51306189);广东省省级科技计划项目(2014A010106022);中国科学院STS计划项目(KFJ-EW-STS-138)
陈伦刚(1981— ),男,湖南祁阳人,副研究员,博士,主要从事生物质催化转化制备液态烃燃料的研究工作
*通讯作者:马隆龙,研究员,博士生导师,研究领域为生物质能源;E-mail: mall@ms.giec.ac.cn。
TQ35;TK6
A
0253-2417(2017)02- 0022-13
陈伦刚,刘勇, 张兴华,等.纤维素催化转化制备C5/C6烷烃燃料的反应与催化体系的研究进展[J].林产化学与工业,2017,37(2):22-34.
猜你喜欢
化工设计通讯(2022年12期)2023-01-24
化工学报(2021年10期)2021-10-31
石油化工(2021年9期)2021-10-18
新能源进展(2021年1期)2021-03-02
化工设计通讯(2020年5期)2020-01-12
石油石化绿色低碳(2019年6期)2019-01-14
天然气技术与经济(2018年1期)2018-03-06
教学考试(高考化学)(2017年3期)2017-08-08
化工设计通讯(2017年6期)2017-03-02
中国塑料(2016年5期)2016-04-16
