
肾上腺外混合性副神经节瘤7例临床病理观察
2017-06-05 15:06吴鸿雁
临床与实验病理学杂志 2017年3期
李 微,付 尧,郑 重,吴鸿雁, 陈 骏
·短篇论著·
肾上腺外混合性副神经节瘤7例临床病理观察
李 微,付 尧,郑 重,吴鸿雁, 陈 骏
目的 探讨混合性副神经节瘤(paraganglioma, PGL)的临床病理特征、免疫表型和分子遗传学改变及其预后的关系。方法 收集7例混合性PGL,采用免疫组化EnVision两步法检测相关免疫组化标志物。结果 7例混合性PGL均与节细胞神经瘤混合,患者年龄21~74岁,平均51岁,男女比为2 ∶5,肿瘤发生于腹膜后、消化道、头颈等部位。CT均示占位性病变,肿瘤边缘清晰,密度不均,可有坏死、出血及囊性变,动脉期呈轻~中度强化,静脉期强化减弱。镜下肿瘤由两种肿瘤细胞构成,一种呈嗜铬细胞分化,细胞排列成不规则巢、团状或腺泡状,以小巢、团状为主,间质血管丰富。细胞大小一致,体积较大,呈多边形,核圆形或卵圆形,可见小核仁,染色质细颗粒状,胞质丰富,粉染,颗粒状,核分裂少见。另一种为束状及编织状排列的施万细胞背景下,散在或聚集分布的神经节细胞,细胞体积大,胞质丰富,核仁清楚,散在分布或排列成群,并可见神经纤维。免疫表型:PGL成分中Syn、CgA均呈弥漫强阳性,而在节细胞神经瘤成分中则显示弱阳性或灶阳性;施万细胞和间质细胞S-100均呈阳性;部分PGL中SDH-B呈灶阳性,部分不表达或弱表达SDH-B;7例Ki-67增殖指数均小于1%。结论 混合性PGL临床非常罕见,其发病部位多位于腹膜后,女性多见,影像学CT示动脉期呈轻~中度强化,静脉期强化减弱,肿瘤密度不均,可有坏死、出血及囊性变。SDH-B表达缺失,提示患者预后不良。
混合性副神经节瘤;SDH-B;预后
副神经节瘤(paraganglioma, PGL)是来源于沿交感神经和副交感神经分布的副神经节肿瘤,可发生于自颅底至盆底的任何靠近中线的部位,包括头颈、胸腔、纵膈、腹腔、盆腔和泌尿生殖道等;肾上腺髓质发生的肿瘤称为嗜铬细胞瘤,肾上腺外的一般称为PGL。文献报道PGL罕见有混合性的存在方式,一般为PGL与节细胞神经瘤混合[1-6],少数情况下可与神经母细胞瘤、节细胞神经母细胞瘤或周围神经鞘膜瘤混合[7-8]。本文现对7例混合性PGL进行分析并结合文献复习,探讨该病的临床病理学特征、诊断及其预后。
1 材料与方法
1.1 材料 收集南京大学医学院附属鼓楼医院2005~2014年诊断手术切除的7例混合性PGL。其中男性2例,女性5例,年龄21~74岁,平均51岁;肿瘤发生于腹膜后4例,颈部、横结肠、十二指肠乳头各1例。3例患者出现下腹部不适、疼痛,4例为体检时发现(表1)。
1.2 方法 所有标本均经10%中性福尔马林固定,常规脱水,石蜡包埋制片,行HE染色及免疫组化EnVision两步法染色。一抗CK、SDH-B、Syn、CgA、S-100、NCAM、CD56、Ki-67,均购自北京中杉金桥公司。具体操作步骤按试剂盒说明书进行。
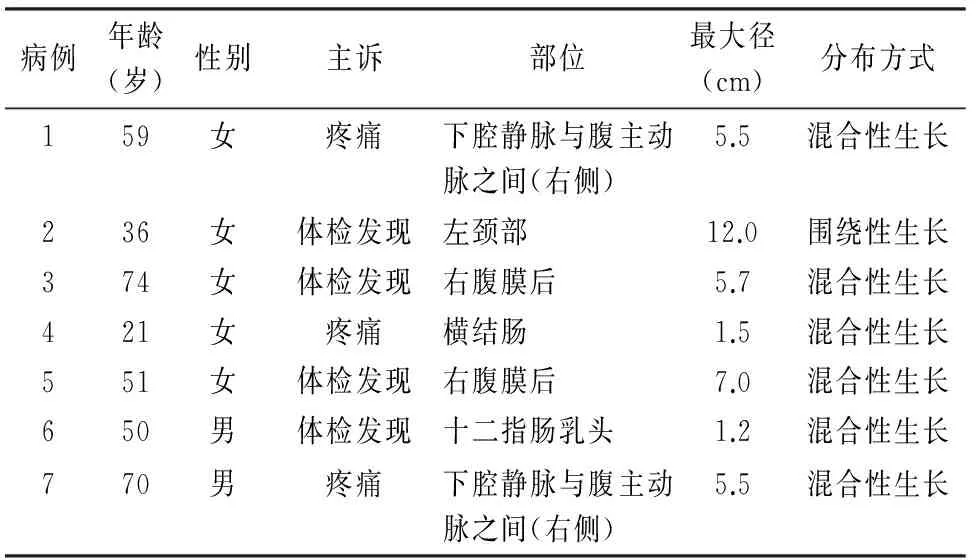
表1 肾上腺外7例混合性PGL临床病理资料
2 结果
2.1 眼观 肿瘤外观呈灰白、灰黄色,表面尚光滑,一般包膜完整,与周围组织分界尚清楚。肿瘤最大径1.2~12 cm,平均值为5.5 cm。肿瘤切面可呈实性、囊实性或囊性,可伴出血、坏死,质中,部分区质硬,切面为灰白、灰黄色,接触空气后部分区域可呈黑色。
2.2 镜检 镜下肿瘤由两种肿瘤细胞构成,一种呈嗜铬细胞分化,细胞排列成不规则巢、团状或腺泡状,以小巢、团状为主,间质血管丰富(图1)。细胞大小一致,体积较大,呈多边形、核圆形或卵圆形,可见小核仁,染色质细颗粒状,胞质丰富,粉染,颗粒状,核分裂少见。另一种为束状及编织状排列的施万细胞背景下,散在或聚集分布的神经节细胞,细胞体积大,胞质丰富,核仁清楚,散在分布或排列成群,并可见神经纤维(图2)。6例肿瘤中上述两种成分表现为相互交织混合分布(图3),1例出现施万细胞围绕嗜铬细胞成分的特殊分布方式(图4)。6例混合性肿瘤未见被膜浸润,1例疑似侵犯被膜,脉管内疑似瘤栓,具有潜在交界性/恶性潜能。
2.3 免疫表型 混合性PGL每种成分的免疫组化与它们正常对应部分或同一类型的肿瘤相似。7例肿瘤中PGL成分CgA(图5)、Syn(图6)均呈弥漫强阳性,而在节细胞神经瘤成分中则显示弱阳性或灶阳性;施万细胞和间质细胞S-100均阳性(图7);7例Ki-67增殖指数均小于1%。两种肿瘤成分显示CK阳性;部分PGL(1/7)中SDH-B阳性,部分不表达(2/7)或呈局灶弱表达(4/7)SDH-B(图8)。
2.4 预后 7例患者均行肿块完整切除,术后随访6~124个月,未见肿瘤复发及转移。
3 讨论
PGL源于有神经嵴交感和副交感链嗜铬组织分布的部位,位于自颅底至骨盆的肾上腺外交感或副交感神经的副神经节,最常见于头颈部、腹部肾血管附近或Zuckerkandl体,后者位于肠系膜下动脉起始部,是肾上腺外最大的嗜铬组织。混合性PGL临床罕见,其典型特征是PGL与节细胞神经瘤、节细胞神经母细胞瘤、神经母细胞瘤或周围神经鞘膜瘤相混合[9]。目前,肾上腺外混合性PGL见少数文献报道[5,10],可发生在腹膜后[1-2,4,11-12]、膀胱[6]、马尾部[13]、胰腺[3,14]、腰背部[5]及椎体[15]等部位,以腹膜后多见。
本组7例肾上腺外混合性PGL有4例发生在腹膜后,发生在消化道2例,头颈1例,混合方式均为PGL与节细胞神经瘤混合。患者年龄21~74岁,平均51岁,男女比为2 ∶5。影像学检查CT均显示为占位性病变,肿瘤边缘清晰,密度不均,可有坏死、出血及囊性变,动脉期呈轻~中度强化,静脉期强化减弱。肿瘤边缘清晰,密度不均;同普通型PGL相比,普通型PGL实性部分呈明显不均匀强化,静脉期则显示持续强化,可与之鉴别。
与PGL相似的是,混合性PGL也可出现功能性,可伴儿茶酚胺和促肾上腺皮质素释放激素(CRH)或其他代谢物的水平升高(如血压升高)。PGL的嗜铬细胞和节细胞神经瘤的节细胞均由神经嵴细胞分化而来,具有分化成体细胞的能力,同时节细胞可产生肽激素和生物胺。因此,混合性PGL中任意一种肿瘤成分均可使激素分泌过多,导致临床症状的出现如头痛、心悸、出汗等。本组7例肾上腺外混合型PGL患者,3例以疼痛为首发症状就诊;4例因体检意外发现占位,其中1例伴高血压症状。另外,文献报道混合性PGL可伴Ⅰ型神经纤维瘤病(NF1)[16]或多发性内分泌肿瘤综合征(MEN 2A)[17],但本组患者均未伴随家族性NF1和(或)MEN 2A。
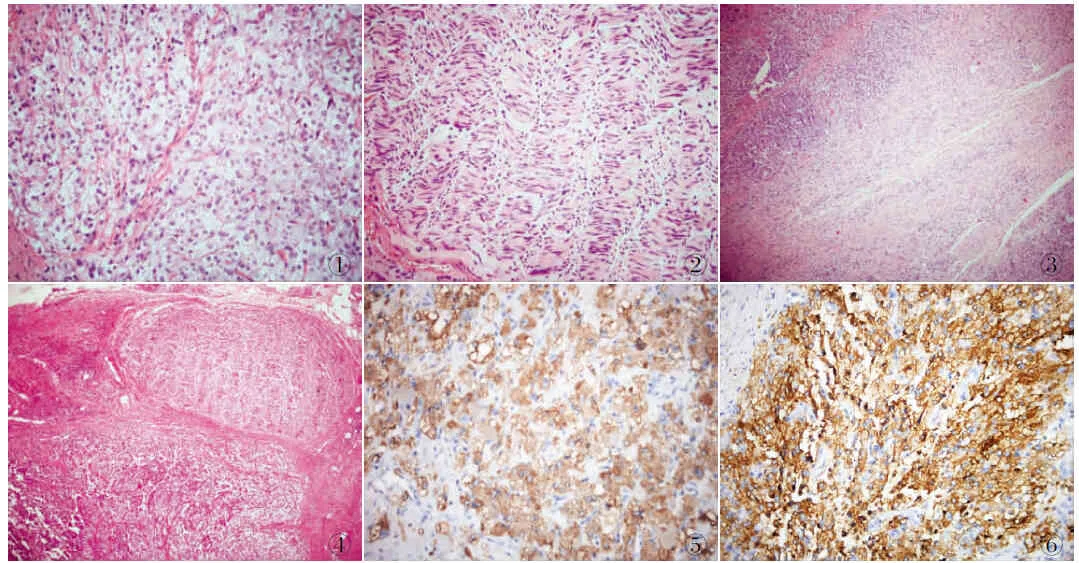

图1 混合性副神经节瘤的嗜铬细胞样成分,肿瘤细胞排列成不规则,呈巢、团状或腺泡状,以小巢、团状为主,间质血管丰富 图2 神经纤维成分中央可见神经节细胞图3 副神经节瘤与神经纤维穿插交织分布 图4 混合性副神经节瘤中施万细胞围绕嗜铬细胞成分的特殊分布方式 图5 混合性副神经节瘤中CgA呈阳性,EnVision两步法 图6 混合性副神经节瘤中Syn呈阳性,EnVision两步法 图7 混合性副神经节瘤中S-100呈阳性,EnVision两步法 图8 混合性副神经节瘤中SDH-B呈阳性,EnVision两步法
混合性PGL常见于成人,平均年龄50岁,无明显性别差异;肿瘤大体可出现补钉状,表现为囊实性。本组患者平均年龄为51岁,女性多见,肿瘤最大径1.2~12 cm,平均值为5.5 cm;肉眼观察肿瘤表面包膜基本完整,与周围组织分界尚清楚。肿瘤切面呈实性、囊实性,伴出血、坏死。混合性PGL镜下表现为PGL和节细胞神经瘤构成,并可见神经纤维。但是两种肿瘤成分排列方式不同,本组6例为PGL与节细胞神经瘤相互交织混合分布,1例表现为节细胞神经瘤围绕PGL分布的独特的分布方式,目前尚未见相关文献报道。
混合性PGL每种成分的免疫组化与它们正常对应部分或同一类型的肿瘤相似。本组混合性PGL成分Syn、CgA 、CD56局面呈弥漫强阳性,而在节细胞神经瘤成分中则显示弱阳性或灶阳性;施万细胞和间质细胞均表达S-100;Ki-67增殖指数小于1%。两种肿瘤成分均显示CK阳性;部分PGL表达SDH-B,部分不表达或弱表达SDH-B。有报道称混合性PGL(与神经母细胞瘤混合)SDH-B呈阴性,可能提示预后不良[8]。研究发现约30%的PGL有胚系RET、Von Hippel-Lindau(VHL)、NF1和SDH-B、SDH-C和SDH-D突变,并且SDH-B在已知有SDH突变的病例中免疫组化染色呈阴性[18]。SDH-B是SDH的一个亚基,SDH-B基因缺失与混合性PGL/神经母细胞瘤相关[8],SDH-B还参与细胞凋亡通路,并在该信号通路中发生胚系突变[19]。另外,Comstock等[7]报道同神经母细胞瘤混合的混合性PGL约50%可出现N-myc扩增,而在经典PGL及其他类型的混合性PGL则均未出现N-myc基因扩增。N-myc基因扩增与1p缺失显著相关,并提示患者预后不良。
大多数单纯性PGL为良性肿瘤,约12%为恶性,目前认为没有单一的组织学特点能独立预测恶性肿瘤的转移倾向;肿瘤融合性坏死、透明滴缺失、原发肿瘤呈粗糙结节状、Ki-67增殖指数高和最大直径>5 cm等,可提示肿瘤具有恶性可能。混合性PGL的生物学行为目前分析较少,已有文献报道其可发生远处转移,发生转移的成分均为不成熟的神经成分[15]。本组6例具有良性生物学行为,1例发现包膜侵犯,但未出现明确转移,因此考虑具有交界性或低度恶性潜能。患者随访6~124个月,均无复发或转移。
[1] Majumder S, Grabska J, Trikudanathan G,etal. Functional ‘composite’ pheochromocytoma-ganglioneuroma presenting as a pancreatic mass[J]. Pancreatology, 2012,12(3):211-214.
[2] Hirasaki S, Kanzaki H, Okuda M,etal. Composite paraganglioma-ganglioneuroma in the retroperitoneum[J]. World J Surg Oncol, 2009,7:81.
[3] Ito H, Kurokawa T, Yokoyama O. Composite paraganglioma with ganglioneuroma in the retroperitoneal space[J]. Int J Urol, 2010,17(4):385-386.
[4] Tohme C A, Mattar W E, Ghorra C S. Extra-adrenal composite pheochromocytoma-ganglioneuroma[J]. Saudi Med J, 2006,27(10):1594-1597.
[5] 胡维维,王 辉,陶金华,等. 肾上腺外副神经节瘤临床病理分析[J]. 临床与实验病理学杂志, 2010,26(1):77-80.
[6] Shankar G M, Chen L, Kim A H,etal. Composite ganglioneuroma-paraganglioma of the filum terminale: case report[J]. J Neurosurgery Spine, 2010,12(6):709-713.
[7] Comstock J M, Willmore-Payne C, Holden J A,etal. Composite pheochromocytoma: a clinicopathologic and molecular comparison with ordinary pheochromocytoma and neuroblastoma[J]. Am J Clin Pathol, 2009,132(1):69-73.
[8] Usuda H, Emura I. Composite paraganglioma-ganglioneuroma of the urinary bladder[J]. Pathol Int, 2005,55(9):596-601.
[9] Dundr P, Dudorkinová D, Povsil C,etal. Pigmented composite paraganglioma-ganglioneuroma of the urinary bladder[J]. Pathol Res Pract, 2003,199(11):765-769.
[10] Lam K Y, Loong F, Shek T W,etal. Composite paraganglioma-ganglioneuroma of the urinary bladder: a clinicopathologic, immunohistochemical, and ultrastructural study of a case and review of the literature[J]. Endocr Pathol, 1998,9(4):353-361.
[11] Linnoila R I, Keiser H R, Steinberg S M,etal. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features[J]. Hum Pathol, 1990,21(11):1168-1180.
[12] Gempt J, Baldawa S S, Weirich G,etal. Recurrent multiple spinal paragangliomas as a manifestation of a metastatic composite paraganglioma-ganglioneuroblastoma[J]. Acta Neurochir (Wien), 2013,155(7):1241-1242.[13] Hu J, Wu J, Li C,etal. Retroperitoneal composite pheochromocytoma-ganglioneuroma: a case report and review of literature[J]. Diagn Pathol, 2013,8(1):63.
[14] Majumder S, Grabska J, Trikudanathan G,etal. Functional ‘composite’ pheochromocytoma-ganglioneuroma presenting as a pancreatic mass[J]. Pancreatology, 2012,12(3):211-214.
[15] Schlisio S, Kenchappa R S, Vredeveld L C W,etal. The kinesin KIF1Bβ acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor[J]. Genes Dev, 2008,22(7):884-893.
[16] Shahani S. Ectopic corticotropin-releasing hormone (CRH) syndrome from metastatic small cell carcinoma: a case report and review of the literature[J]. Diagn Pathol, 2010,5(1):56.
[17] Pytel P, Krausz T, Wollmann R,etal. Ganglioneuromatous paraganglioma of the cauda equina-a pathological case study[J]. Hum Pathl, 2005,36(4):444-446.
[18] Kimura N, Watanabe T, Fukase M,etal. Neurofibromin and NF1 gene analysis in composite pheochromocytoma and tumors associated with von recklinghausen’s disease[J]. Mod Pathol, 2002,15(3):183-188.
[19] Gullu S, Gursoy A, Erdogan M F,etal. Multiple endocrine neoplasia type 2A/localized cutaneous lichen amyloidosis associated with malignant pheochromocytoma and ganglioneuroma[J]. J Endocrinol Invest, 2005,28(10):734-737.
时间:2017-3-16 14:23
http://kns.cnki.net/kcms/detail/34.1073.R.20170316.1423.016.html
国家青年科学基金(81502170)、中央高校基本科研业务费专项资金(021414380101)
南京大学医学院附属鼓楼医院病理科,南京 210008
李 微,女,硕士,医师。E-mail: leeweilw@hotmail.com 陈 骏,男,副主任医师,通讯作者。E-mail: ichenjun@qq.com
R 737.11
B
1001-7399(2017)03-0307-03
10.13315/j.cnki.cjcep.2017.03.016
接受日期:2017-02-05
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
临床肝胆病杂志(2020年9期)2020-09-28
人物画报(2020年28期)2020-03-09
人物画报(2020年10期)2020-01-05
传感器世界(2019年6期)2019-09-17
中国临床医学影像杂志(2019年5期)2019-08-27
中医眼耳鼻喉杂志(2019年2期)2019-04-13
现代泌尿生殖肿瘤杂志(2018年5期)2018-11-29
健康管理(2016年6期)2016-05-14
飞碟探索(2016年5期)2016-05-10
